Muchas personas no entienden por qué o cómo las personas se vuelven adictas a las drogas. Ellos pueden erróneamente pensar que aquellos que usan drogas les faltan principios morales o fuerza de voluntad y que ellos pueden dejar de usar drogas simplemente tomando la decisión de parar. En realidad, la drogadicción es una enfermedad compleja, y dejar de usar las drogas usualmente requiere más que buenas intenciones o una gran voluntad. Las drogas cambian el cerebro en maneras que hacen que el dejar de usarlas sea duro, hasta para aquellos que quieren dejarlas. Afortunadamente, los científicos conocen más que nunca cómo las drogas afectan al cerebro y han encontrado tratamientos que pueden ayudar a las personas a recuperarse de la drogadicción y llevarlos a tener vidas productivas.
¿Qué es la drogadicción?
La drogadicción es una enfermedad crónica caracterizada por la búsqueda y el uso compulsivo e incontrolable de una droga, a pesar de las consecuencias adversas. Para la mayoría de las personas, la decisión inicial de usar drogas es voluntaria, pero el uso repetido de las drogas puede llevar a cambios en el cerebro que desafían el autocontrol de una persona adicta e interfiere con su habilidad de resistir los deseos intensos de usar drogas. Estos cambios del cerebro pueden ser persistentes, por lo cual se considera la drogadicción una enfermedad «reincidente»—las personas en recuperación del trastorno del uso de drogas están a un alto riesgo a volver a usar drogas, aunque lleven años sin usarlas.
Es común que una persona recaiga, pero la recaída no significa que el tratamiento no sirva. Similar a otras condiciones crónicas de la salud, el tratamiento debe de ser continuo y ajustado basado en cómo el paciente responda. Los planes de tratamiento necesitan ser revisados a menudo y modificados para adaptarse a las necesidades cambiantes del paciente.
¿Qué pasa con el cerebro cuando una persona usa drogas?
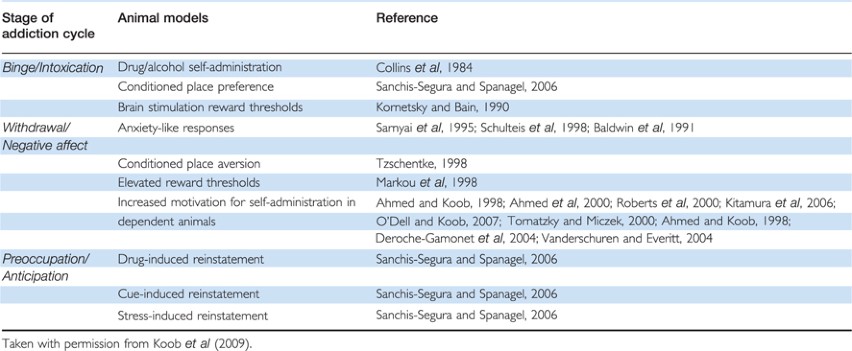
La mayoría de las drogas afectan al circuito de recompensa del cerebro, inundándolo con el químico mensajero la dopamina. Este sistema de recompensa controla la habilidad del cuerpo de sentir placer y motiva a la persona a repetir comportamientos necesitados para prosperar, como comer y pasar tiempo con los seres queridos. Esta sobre estimulación del circuito de recompensa causa la intensa recompensa placentera que puede llevar a las personas a tomar drogas una y otra vez.
Cuando una persona continúa consumiendo drogas, el cerebro se ajusta al exceso de la dopamina al producir menos de ella, y/o reduce la habilidad de las células en el circuito de recompensa a responderle. Esto reduce la recompensa que la persona siente, comparado a la recompensa que sintieron cuando tomaron la droga por primera vez—un efecto conocido como la tolerancia. Ellos pueden usar más de la droga, tratando de alcanzar al mismo nivel de recompensa de la dopamina. También puede causarles sentir menos placer de otras cosas que alguna vez disfrutaban, como la comida y actividades sociales.
El uso de drogas a largo plazo también causa cambios en otros sistemas químicos y circuitos del cerebro, afectando las siguientes funciones:
aprendizaje
criterio
capacidad de tomar decisiones
estrés
memoria
comportamiento
A pesar de estar conscientes de estos efectos perjudiciales, muchas personas que usan drogas continúan a usarlas, lo que es la característica de la adicción.
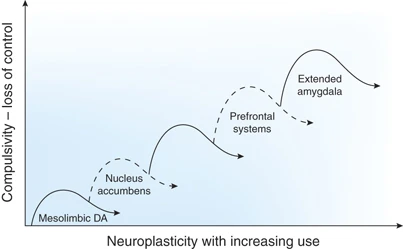
El circuito de recompensa del cerebro
¿Por qué es que algunas personas se vuelven adictas a las drogas y otras no?
No hay un solo factor que puede predecir si una persona se volverá adicta a las drogas. Una combinación de factores influye el riesgo a la adicción. Si una persona tiene más factores de riesgo, el potencial de llevarse adicta con usar drogas es más grande. Por ejemplo:
Biología. Los genes con los que una persona nace cuentan por la mitad de los riesgos para la adicción. Género, etnicidad y la presencia de otros trastornos mentales pueden también influir el riesgo del uso de drogas y la adicción.
Ambiente. El ambiente de una persona incluye varias influencias diferentes, de su familia y sus amigos hasta el estado económico y su calidad de vida en general. Factores como la presión social, abuso físico y sexual, exposición temprana a las drogas, estrés y orientación parental pueden afectar mucho la probabilidad de que una persona use drogas y que lleve a la adicción.
Desarrollo. Factores genéticos y ambientales interactúan con etapas críticas del desarrollo de una persona en su vida que afectan el riesgo de adicción. Aunque usar drogas a cualquier edad puede llevar a la adicción, lo más joven que uno empieza a usar drogas, lo más probable que progrese a la adicción. Esto es particularmente problemático para los adolescentes. Ya que todavía se están desarrollando las áreas del cerebro que controlan la capacidad de tomar decisiones, el criterio y el autocontrol, los adolescentes en especial pueden estar propensos a comportamientos riesgosos incluyendo el probar de las drogas.
¿Puede ser prevenida o curada la drogadicción?
El tratamiento para la drogadicción, similar a otras enfermedades crónicas, como la diabetes, asma o cardiopatía, generalmente no indica una cura. Sin embargo, la adicción es tratable y puede ser exitosamente manejada. Las personas que se están recuperando de una adicción estarán en riesgo de experimentar una recaída por años y posiblemente por toda la vida. Las investigaciones demuestran que combinar los medicamentos con terapias conductuales para tratar la adicción asegura la mejor posibilidad de éxito para la mayoría de los pacientes. Los enfoques de tratamiento adaptados a la forma del uso de drogas de cada paciente y a cualquier problema médico, mental y social ocurriendo simultáneamente pueden llevar a recuperación continua.
Otra buena noticia es que el uso de drogas y la adicción son prevenibles. Los resultados de las investigaciones patrocinadas por NIDA han demostrado que los programas de prevención involucrando familias, escuelas, comunidades y medios de comunicación son eficaces para prevenir o reducir el uso de drogas y la adicción. Aunque eventos personales y factores culturales afectan las tendencias del uso de las drogas, cuando las personas jóvenes perciben el uso de drogas como riesgoso, ellos tienden a reducir el uso de drogas. Por lo tanto, la educación y la divulgación son claves con ayudar que las personas entiendan los posibles riesgos del uso de drogas. Los profesores, los padres y los proveedores de salud tienen roles claves en educar a los jóvenes y prevenir el uso de drogas y la adicción.
PUNTOS PARA RECORDAR:
La drogadicción es una enfermedad crónica caracterizada por la búsqueda y el uso compulsivo e incontrolable de una droga, a pesar de las consecuencias adversas.
Cambios en el cerebro que ocurren con el tiempo con el uso de drogas desafían el autocontrol de una persona adicta e interfieren con su habilidad de resistir los deseos intensos de usar drogas. Esto es por qué la drogadicción es también considerada una enfermedad reincidente.
La recaída es el volver a usar drogas después de haber tratado de parar. La recaída indica la necesidad por más o diferente tratamiento.
La mayoría de las drogas afectan al circuito de recompensa del cerebro, inundándolo con el químico mensajero la dopamina. Esta sobre estimulación del circuito de recompensa causa la intensa recompensa placentera que lleva a las personas a tomar las drogas una y otra vez.
Con el tiempo, el cerebro se ajusta al exceso de la dopamina, lo que reduce la recompensa que la persona siente, comparado a la recompensa que sintieron cuando tomaron la droga por primera vez—un efecto conocido como la tolerancia. Ellos pueden tomar más de la droga, tratando de alcanzar al mismo nivel de recompensa de la dopamina.
No hay un solo factor que puede predecir si una persona se volverá adicta a las drogas. Una combinación de factores genéticos, ambientales y desarrollantes influyen el riesgo a la adicción. Si una persona tiene más factores de riesgo, el potencial de llevarse adicta con usar drogas es más grande.
La drogadicción es tratable y puede ser exitosamente manejada.
Otra buena noticia es que el uso de drogas y la adicción son prevenibles. Los profesores, los padres y los proveedores de salud tienen roles claves en educar a los jóvenes y prevenir el uso de drogas y la adicción.
Para más información
Para más información sobre entender el uso de drogas y la adicción, visite:
La adicción no es sólo una cuestión de fuerza de voluntad. Es una enfermedad crónica del cerebro, dice una nueva definición destinada a ayudar a las familias y a los médicos a comprender mejor los retos que supone su tratamiento.
«La adicción es mucho más que el mal comportamiento de las personas», dice el Dr. Michael M. Miller, de la Sociedad Americana para la Medicina de la Adicción.
Eso es cierto ya sea
que se trate de drogas y alcohol o de juegos de azar y comidas compulsivas,
dijo el lunes el grupo de médicos. Y al igual que otras enfermedades
crónicas como la enfermedad cardíaca o la diabetes, el tratamiento de la
adicción y la prevención de recaídas es un esfuerzo a largo plazo, concluyeron
los especialistas.
La adicción generalmente se describe por sus síntomas de
comportamiento: los niveles altos, los antojos y las cosas que las personas
harán para lograr uno y evitar el otro. La nueva definición no está en
desacuerdo con la guía estándar para el diagnóstico basada en esos síntomas.
Pero dos décadas de neurociencia han descubierto cómo la
adicción secuestra diferentes partes del cerebro para explicar qué es lo que
incita esos comportamientos y por qué pueden ser tan difíciles de superar. La
declaración de política de la sociedad, publicada en su sitio web, no es una
dirección nueva sino parte de un esfuerzo por traducir esos hallazgos a los
médicos de atención primaria y al público en general.
«El problema de comportamiento es el resultado
de una disfunción cerebral«, concuerda la Dra. Nora Volkow, directora
del Instituto Nacional sobre el Abuso de Drogas.
Dio la bienvenida a la declaración como una manera de ayudar al trabajo de su propia agencia para alentar a más médicos de atención primaria a que examinen a sus pacientes en busca de signos de adicción. NIDA estima que 23 millones de estadounidenses necesitan tratamiento por abuso de sustancias, pero solo unos 2 millones obtienen esa ayuda. Tratando de agregar compasión a los hallazgos del cerebro, NIDA incluso ha hecho que las lecturas de «El largo día del viaje hacia la noche» de Eugene O’Neill sean una parte de las reuniones donde los médicos de atención primaria aprenden sobre la adicción.
Luego está la frustración de las recaídas, que los médicos y las familias deben saber que son comunes para una enfermedad crónica, dice Volkow.
«Tienes miembros de la familia que dicen: ‘Está bien, has estado en un programa de desintoxicación, ¿por qué estás tomando drogas?'», Dice ella. «La patología en el cerebro persiste durante años después de haber dejado de tomar el medicamento».
¿Qué pasa en el cerebro? Es un juego complejo de redes emocionales, cognitivas y de comportamiento.
La genética desempeña un papel, lo que significa que algunas personas son más vulnerables a una adicción si, por ejemplo, experimentan con drogas en la adolescencia o terminan con analgésicos potentes recetados después de una lesión.
La edad también lo hace. La corteza frontal ayuda a frenar los comportamientos poco saludables, explica Volkow. Es donde el lado del razonamiento del cerebro se conecta con las áreas relacionadas con la emoción. Es una de las últimas regiones neuronales en madurar, una de las razones por las que es más difícil para un adolescente soportar la presión de sus compañeros para experimentar con drogas.
Incluso si no eres biológicamente vulnerable para empezar, tal vez pruebes alcohol o drogas para enfrentar un ambiente estresante o doloroso, dice Volkow.
Cualquiera que sea la razón, el sistema de recompensa del cerebro puede cambiar a medida que una sustancia química llamada dopamina lo condiciona a rituales y rutinas que están vinculados a la obtención de algo que le ha resultado placentero, ya sea un paquete de cigarrillos o unas copas o incluso comer en exceso. Cuando alguien es realmente adicto, ese sistema deformado le hace volver incluso después de que el cerebro se haya acostumbrado tanto al subidón que ya no es placentero.
No se equivoquen: los pacientes aún deben seguir eligiendo defenderse y tratar una adicción, subraya Miller, director médico del Centro de Recuperación de Herrington en el Rogers Memorial Hospital en Oconomowoc, Wisconsin.
Pero comprender algunas de las reacciones cerebrales en la raíz del problema «con suerte reducirá parte de la vergüenza de algunos de estos problemas, con suerte reducirá el estigma», dice.
Y mientras que la mayoría de las neurociencias se centran en la adicción a las drogas y el alcohol, la sociedad señala que es posible volverse adicto al juego, el sexo o la comida. aunque no hay buenos datos sobre la frecuencia con la que sucede. Es hora de que un mejor estudio lo averigüe, dice Miller.
Mientras tanto, Volkow dice que se está llevando a cabo una investigación intrigante para utilizar esos hallazgos cerebrales para desarrollar mejores tratamientos, no solo para bloquear temporalmente la actividad de un adicto, sino también para fortalecer los circuitos cerebrales subyacentes para evitar la recaída.
Encabezando la lista de deseos de Miller: Aprender por qué a algunas personas les resulta más fácil y rápida la recuperación que a otras, y «cómo es la curación del cerebro».
Uno de los descubrimientos más notables de las neurociencias ha sido la determinación de los circuitos de recompensa. Se sabe que Fedor Dostoievski escribió una de sus novelas más reconocidas, El jugador, acosado por las deudas, el apasionamiento amoroso y el desatino. Se suele ver el reflejo de esa pesadumbre en Alexei Ivánovich, el protagonista de la novela, un hombre seducido por la bella Polina, pero también por el juego. Tanto, que en los últimos párrafos se confiesa diciendo: “Si pudiera dominarme durante una hora, sería capaz de cambiar mi destino”.
Esta frase permite definir de manera categórica de qué
hablamos cuando hablamos de adicción, una forma particularmente peligrosa de
búsqueda de placer.
La adicción fue considerada durante mucho tiempo como una debilidad moral o una falta de fuerza de voluntad. Por el contrario, actualmente es reconocida como una enfermedad crónica con cambios cerebrales específicos. Así como la enfermedad cardíaca afecta el corazón y la hepatitis, el hígado, la adicción afecta el cerebro, lo secuestra.
La palabra “adicción” deriva del latín “esclavizado por” y se manifiesta en el anhelo por el objeto del que se es adicto, la pérdida de control sobre su uso y la necesidad imperiosa de continuar así a pesar de las consecuencias adversas que eso conlleva
Durante muchos años se creía que sólo el alcohol y las drogas podían causar adicción. Investigaciones recientes han demostrado que ciertas actividades como el juego, las compras, el sexo, la comida e, incluso, la tecnología, también pueden cooptar el cerebro y son registrados por éste en forma similar a las drogas y el alcohol. El consenso científico actual sugiere que estos placeres pueden representar múltiples expresiones de un proceso cerebral común subyacente.
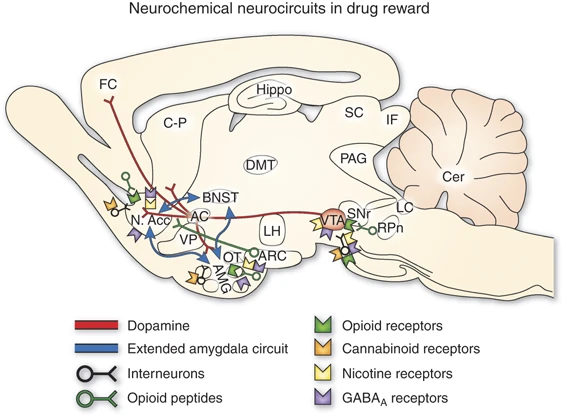
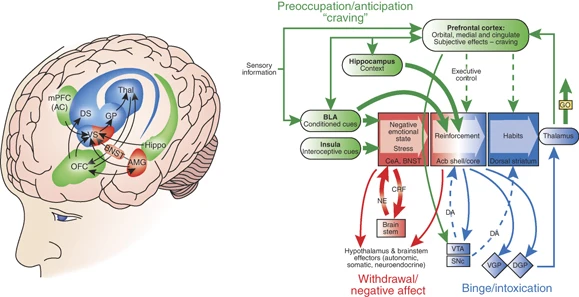
Uno de los descubrimientos más notables de las neurociencias ha sido la determinación de los circuitos de recompensa. Se trata de mecanismos de placer que involucran diferentes regiones cerebrales que se encuentran comunicados mediante mensajeros químicos llamados “neurotransmisores”. La dopamina es un mensajero químico involucrado en la motivación, el placer, la memoria y el movimiento, entre otras funciones. En el cerebro, el placer se produce a través de la liberación de la dopamina en el “núcleo accumbens”, una región a la cual los neurocientíficos llaman el “centro de placer del cerebro”. Justamente la acción de una droga adictiva funciona a partir de la influencia en ese sistema.
Como sabemos, algunos adictos llegan a focalizarse en conseguir y consumir la droga excluyendo todos los demás aspectos de sus vidas: descuidan a su familia, su trabajo, su propia salud. A sabiendas de que se están destruyendo a sí mismos, siguen con el consumo de la droga y, a medida que continúan con su uso, se hacen tolerantes. Así, las dosis que inicialmente utilizaron para estimularse ya no son eficaces y necesitan usar una dosis más alta. En la década de 1950, dos psicólogos canadienses, James Olds y Peter Milner, hicieron unos experimentos muy famosos en los cuales implantaron electrodos en el cerebro basal de las ratas y descubrieron que las drogas adictivas pueden liberar de dos a diez veces -y de forma más rápida- la cantidad de dopamina que las recompensas naturales.
Antes se pensaba que la experiencia del placer era suficiente para inducir a la gente a seguir buscando una sustancia adictiva. Pero nuevas investigaciones sugieren que la situación es más compleja. La dopamina no sólo contribuye a la experiencia del placer, sino que también desempeña un papel en el aprendizaje y la memoria, dos elementos clave en la transición de consumir algo a convertirse en adicto. La investigadora Nora Volkow, en Estados Unidos, utilizó una técnica de neuroimágenes denominada “tomografía por emisión de positrones” para etiquetar los receptores de dopamina en el cerebro humano y descubrió que efectivamente el funcionamiento normal del sistema dopaminérgico cerebral parece estar afectado en el abuso crónico de drogas. Sin embargo, este estudio planteó preguntas fundamentales a partir de esa conclusión: ¿Son estos cambios en los receptores dopaminérgicos de los consumidores de drogas las consecuencias del abuso en el consumo o es el abuso de drogas una consecuencia de una predisposición biológica, lo que quiere decir que estos cambios en los receptores dopaminérgicos están antes del consumo de drogas?
Otro enigma recurrente es el que plantea el comportamiento, a menudo impulsivo, de algunos consumidores de drogas. Nuevamente se evidencia la pregunta sobre cuál es la causa y cuál es el efecto. La vulnerabilidad genética contribuye al riesgo de desarrollar una adicción. Los estudios de gemelos y adopción muestran que alrededor del 40% al 60% de la susceptibilidad a la adicción es hereditaria. Pero el comportamiento juega un papel clave, especialmente cuando se trata de reforzar un hábito. Cada uno de nosotros tiene que tomar decisiones acerca de si realizamos algo que queremos hacer o no (por ejemplo, desear comer un chocolate pero no hacerlo para evitar consecuencias negativas en el mediano plazo). A veces esto no se puede controlar, pero son más las veces que uno puede. En las personas que son adictas, como vimos en el personaje de El jugador, este control es muy difícil.
En los comportamientos compulsivos fallan los frenos del cerebro, aquellos que deberían ejercer el control cognitivo
La persona que es adicta no quiere serlo. Su adicción ya le costó su trabajo, su pareja, su bienestar. Sin embargo, no puede resistir la tentación. Como dijimos al principio, se trata de una enfermedad de la que actualmente no existe cura. Se la debe tratar como otras enfermedades crónicas (hipertensión, asma, cáncer) y, como tal, mantener el tratamiento ya que, de otro modo, el paciente recae.
La adicción se aprende y se almacena como memoria en el cerebro, por lo que la recuperación es un proceso lento. Incluso después de que una persona renuncia, por ejemplo, al consumo de drogas, durante semanas, meses, e incluso, años, la exposición al sitio de la droga, caminar por una calle donde la compraron o tropezar con personas que siguen consumiendo les trae un tremendo impulso a querer consumir de nuevo. Existe una serie de tratamientos que lograron eficacia, por lo general al combinar estrategias de autoayuda, de psicoterapia y de rehabilitación. Para algunos tipos de adicciones, ciertos medicamentos también pueden ayudar.
En una carta de mayo de 1867, el propio Dostoievski -no ya su personaje- le cuenta mortificado a su esposa que todo el dinero con el que contaba lo ha perdido en el casino. Así le describe el escritor ruso su derrotero: “Al principio perdí muy poco, pero cuando comencé a perder, sentía deseos de desquitar lo perdido y cuando perdí aún más, ya fue forzoso seguir jugando para recuperar aunque sólo fuera el dinero necesario para mi partida, pero también eso lo perdí.” Y le promete para el futuro: “De hoy en adelante voy a trabajar, voy a trabajar y voy a demostrar de qué soy capaz.”
El mismo desaliento y el mismo propósito de enmienda de todos cuando lo que no se puede es dominarse y, de este modo, cambiar el destino. Así y todo, pudo cumplir con eso de escribir y demostrar de todo lo que era capaz.
Facundo Manes, es neurólogo, neurocientífico y político argentino creador del Instituto de Neurología Cognitiva (INECO), presidente de la Fundación INECO, ex rector de la Universidad Favaloro y director del Instituto de Neurociencias de la Fundación Favaloro
Las benzodiazepinas se recetan ampliamente para una variedad de afecciones, especialmente la ansiedad y el insomnio. Son relativamente seguros y, con una sobredosis, rara vez resultan en la muerte. Sin embargo, usadas crónicamente, las benzodiazepinas pueden ser adictivas. Estos agentes a menudo se toman en combinación con otras drogas de abuso por parte de pacientes con trastornos de adicción. En tales pacientes, las alternativas a las benzodiazepinas pueden ser preferibles y pueden incluir antidepresivos, anticonvulsivos, buspirona, agentes antihipertensivos y los nuevos medicamentos neurolépticos. Se debe tener precaución al prescribir benzodiazepinas a pacientes con un historial actual o remoto de abuso de sustancias.
Existe poca duda sobre la eficacia terapéutica de las
benzodiazepinas para reducir la ansiedad, inducir el sueño y sofocar los
síntomas de pánico. Como se señaló en un informe de 1990 de la American Psychiatric Association (APA)
sobre la dependencia, toxicidad y abuso de las benzodiazepinas,1 la
eficacia ansiolítica e hipnótica de las benzodiazepinas ha sido bien
establecida por numerosos estudios controlados con placebo.
Las benzodiazepinas se recetan ampliamente, con cuatro de ellas, alprazolam (Xanax), clonazepam (Klonopin), diazepam (Valium) y lorazepam (Ativan), que figuran entre los 100 medicamentos más recetados con mayor frecuencia.2 Las benzodiazepinas generalmente producen efectos casi inmediatos y, por lo tanto, pueden prescribirse para uso a corto plazo, intermitente, «según sea necesario». Debido a que muchos de los trastornos de ansiedad aumentan y disminuyen con el tiempo, los pacientes con estos trastornos a menudo prefieren las benzodiazepinas porque estos agentes pueden tomarse de manera intermitente, cuando los pacientes sienten la necesidad de tomarlos, y la mayoría de los pacientes pueden usar las benzodiazepinas con prudencia.1
Las benzodiazepinas también se recetan ampliamente por otras razones, como espasticidad muscular, trastornos convulsivos, sedación prequirúrgica, trastornos del movimiento involuntario, desintoxicación del alcohol y otras sustancias, y ansiedad asociada con afecciones cardiovasculares o gastrointestinales 3 (Tabla 1)
Tabla 1 Usos clínicos de las benzodiazepinas -Desórdenes de ansiedad · Ansiedad aguda · Trastorno de ansiedad generalizada -Trastorno de pánico -Fobias (sociales, simples) -Trastorno de estrés postraumático -Desorden obsesivo compulsivo -Insomnio -Ansiedad asociada a la enfermedad médica. · Cardiovascular · Gastrointestinal · Trastorno somatomorfo -Trastornos convulsivos -Estado epiléptico agudo · Convulsiones neonatales o convulsiones febriles. -Preeclampsia -Tétanos -Adjunto a otros anticonvulsivos -Amnestico (antes de la cirugía o procedimiento) -Trastornos espásticos y otros tipos de espasmos musculares agudos. · Parálisis cerebral · Esclerosis múltiple · Paraplejia secundaria a traumatismo espinal -Trastornos del movimiento involuntario. · Síndrome de la pierna inquieta · Acatisia asociada al uso neuroléptico · Trastornos coreiformes · Mioclonía -Desintoxicación del alcohol y otras sustancias -Agitación o ansiedad asociada a otros trastornos psiquiátricos · Manía aguda · Enfermedad psicótica · Ansiedad asociada a la depresión · Trastornos de control de impulso -Catatonia o mutismo -Otros usos complementarios · Cirugía · Odontología · Estudios de diagnóstico, como tomografía computarizada, resonancia magnética y endoscopia · Cardioversión · Quimioterapia
Información de Hollister L, Muller-Oerlinghausen B, Rickels K, Shader R. Usos clínicos de los benzodiacepinas. J Clin Psychopharmacol 1993; 13 (supl. 1): 1–169 .
De acuerdo con el informe de la APA sobre las benzodiazepinas,1 de 11 a 15 por ciento de la población adulta ha tomado una benzodiazepina una o más veces durante el año anterior, pero solo 1 a 2 por ciento ha tomado benzodiazepinas diariamente durante 12 meses o más. Sin embargo, en entornos de tratamiento psiquiátrico y en poblaciones con abuso de sustancias, la prevalencia del uso, abuso y dependencia de las benzodiazepinas es sustancialmente mayor que en la población general. 4, 5
Debido a que las benzodiazepinas son sustancias controladas con potencial de abuso, se debe prestar atención especial al historial de adicciones del paciente antes de que se receten estos agentes. Una comprensión de la toxicidad y los efectos secundarios de las benzodiazepinas, los patrones de abuso y los agentes ansiolíticos e hipnóticos alternativos puede ayudar a los médicos a maximizar los resultados del tratamiento y reducir los riesgos de responsabilidad médico-legal.
Neuroquímica
Los receptores de benzodiazepinas son ubicuos en todo el sistema nervioso central. Los receptores de benzodiazepina están vinculados predominantemente a los receptores de ácido amino butírico (GABA), que sensibilizan los receptores de benzodiazepina al neurotransmisor GABA, el neurotransmisor inhibitorio más prominente en el sistema nervioso central. Las benzodiazepinas aumentan la afinidad del sitio de reconocimiento de GABA al inducir cambios conformacionales que hacen que la unión de GABA sea más eficaz. La activación del complejo benzodiazepina-GABA-cloruro de ionófora es responsable de producir los efectos ansiolíticos terapéuticos de las benzodiazepinas y de mediar en muchos de los efectos secundarios y, posiblemente, de la dependencia y la abstinencia de estos fármacos. 6
De manera similar, otros sitios para la unión de fármacos y neurotransmisores están asociados con el complejo del receptor GABA, que sirve como un sitio de acción primario de las benzodiazepinas, barbitúricos y otros hipnóticos sedantes, como el alcohol.6 Las benzodiazepinas y los barbitúricos actúan en sitios de unión separados en el receptor para potenciar la acción inhibitoria de GABA. Lo hacen alterando de forma alostérica el receptor (cambiando su conformación) para que tenga una mayor afinidad de unión por GABA. El etanol modifica el receptor al alterar el entorno de la membrana para que tenga una mayor afinidad por el GABA y los otros fármacos sedantes-hipnóticos. Que las benzodiazepinas, los barbitúricos y el etanol tienen acciones relacionadas con un tipo de receptor común, lo que explica su sinergia farmacológica y la tolerancia cruzada. Por lo tanto, las benzodiazepinas se utilizan durante la desintoxicación del alcohol.
Con el uso prolongado de dosis altas de benzodiazepinas (o etanol), hay una disminución aparente en la eficacia de los receptores GABA-A, probablemente un mecanismo de tolerancia.6,7 Cuando las benzodiazepinas en dosis altas o el etanol se suspenden abruptamente, este estado «regulado a la baja» de la transmisión inhibitoria se desenmascara, lo que lleva a síntomas de abstinencia característicos como ansiedad, insomnio, hiperactividad autonómica y, posiblemente, convulsiones.
Toxicidad y efectos secundarios
Con la introducción de clordiazepóxido (Librium) en 1960, y debido a la relativa seguridad de las benzodiazepinas, estos agentes reemplazaron rápidamente a los barbitúricos como hipnóticos sedantes. Causan significativamente menos depresión respiratoria que los barbitúricos y, en consecuencia, rara vez son letales en una sobredosis.
Como clase de fármacos, las benzodiazepinas comparten muchas propiedades clínicas, aunque los diferentes agentes de esta clase pueden mostrar diferentes propiedades farmacocinéticas y farmacodinámicas (Tabla 2). Las propiedades farmacológicas, como la potencia, la vida media y la lipofilicidad, la duración del tratamiento y la tasa de aumento o disminución de la dosis influyen en la aparición de efectos secundarios.1 El desarrollo de la dependencia fisiológica es algo predecible y es proporcional a la exposición total a las benzodiazepinas (dosis x duración del tratamiento), aunque puede existir una variabilidad significativa entre los pacientes.
Tabla 2 Potencia y vida media de varias benzodiazepinas
Benzodiacepinas de alta potencia
Medicamentos con una vida media corta. · Alprazolam (Xanax) · Lorazepam (Ativan) · Triazolam (Halcion)
Medicamentos con una larga vida media · Clonazepam (Klonopin)
Benzodiacepinas de baja potencia
Medicamentos con una vida media corta. · Oxazepam (Serax) · Temazepam (Restoril)
Medicamentos con una larga vida media · Clordiazepóxido (Librium) · Clorazepate (Tranxene) · Diazepam (valium) · Flurazepam (Dalmane)
Toxicidad en interacciones de medicamentos
Cuando se usan solas, las benzodiazepinas tienen un riesgo extremadamente bajo de toxicidad aguda. Sin embargo, las benzodiazepinas a menudo se usan con otros tipos de medicamentos, incluidas otras drogas con potencial de abuso, y estas drogas pueden aumentar los efectos tóxicos de las benzodiazepinas. Estos últimos interactúan de forma sinérgica con otros depresores del sistema nervioso central, incluidos otros hipnóticos, antidepresivos sedantes, neurolépticos, anticonvulsivos, antihistamínicos y alcohol.8 Las sobredosis fatales en pacientes adictos a menudo involucran la combinación de benzodiazepinas y alcohol, con o sin opiáceos. Además, pueden producirse interacciones farmacocinéticas farmacológicas. Por ejemplo, los inhibidores selectivos de la recaptación de serotonina (ISRS) pueden aumentar los niveles sanguíneos de diazepam,9 y la nefazadona (Serzone) puede aumentar los niveles de alprazolam10 a través de la inhibición de la enzima hepática, lo que lleva a un aumento de los efectos sedantes-hipnóticos o efectos secundarios.
Retardo psicomotor
La desaceleración psicomotora puede ser especialmente profunda después de la administración inicial de una benzodiazepina o con un aumento repentino de la dosis. También se puede observar en pacientes, como los ancianos, que tienen tasas disminuidas de metabolismo o mayor susceptibilidad a la depresión del sistema nervioso central.8 Los síntomas psicomotores incluyen somnolencia, falta de concentración, ataxia, disartria, falta de coordinación motora, diplopía, debilidad muscular, vértigo y confusión mental.11 Los estudios de los efectos psicomotores sugieren que las benzodiazepinas retrasan el tiempo de reacción y afectan las habilidades de manejo, lo que aumenta el riesgo de accidentes automovilísticos en pacientes que toman estos agentes.12
Deterioro de la memoria
Las benzodiazepinas inducen amnesia anterógrada, lo que explica los efectos beneficiosos de las benzodiazepinas como el midazolam (Versed) para la medicación prequirúrgica. Estos efectos amnésicos específicos parecen estar separados de la sedación.11 La memoria episódica (el recuerdo de eventos recientes y las circunstancias en que ocurrieron y sus secuencias de tiempo) está particularmente deteriorado y más notablemente en los bebedores de alcohol que también usan benzodiazepinas. Las deficiencias específicas en la capacidad visuoespacial y la atención sostenida también se han descrito en pacientes que han tomado dosis terapéuticas de benzodiazepinas regularmente durante más de un año.13
Desinhibición pardógica
El aumento de la excitación, la irritabilidad, la agresión, la hostilidad y la impulsividad pueden ocurrir en algunos pacientes que toman benzodiacepinas. Esta desinhibición paradójica puede, en casos raros, resultar en ataques de rabia o violencia, u otras conductas indiscrecionales o antisociales.14 Tales reacciones pueden deberse a la desinhibición de las tendencias de comportamiento normalmente suprimidas por las restricciones sociales (como también puede ocurrir con el alcohol). Estas reacciones ocurren con mayor frecuencia en niños, ancianos y personas con discapacidades del desarrollo.
Depresión a apoyo emocional
Se ha observado una asociación entre el uso de benzodiazepinas y los síntomas depresivos y, en algunos casos, la aparición de ideas suicidas. Algunas pruebas indican que las dosis más altas de benzodiazepinas se asocian con un mayor riesgo de depresión y que reducir la dosis o interrumpir la terapia puede resolver los síntomas depresivos.15 Aunque el mecanismo de esta acción no está claro, la depresión relacionada con las benzodiazepinas podría ocurrir como resultado fisiológico de una reducción en la actividad de la monoamina central.
La «anestesia emocional» también se puede ver en la práctica clínica. Este efecto puede ser buscado por drogadictos que se vuelven progresivamente más incapaces de tolerar sus emociones y factores estresantes de la vida.
Efectos adversos en el embarazo
Las benzodiazepinas atraviesan la placenta y se clasifican como teratógenos de clase D. Pueden conducir al desarrollo de la dependencia y los síntomas de abstinencia consecuentes en el feto.16 Las benzodiazepinas se excretan en la leche materna y, por lo tanto, suelen estar contraindicadas en las madres que amamantan.
Tolerancia
La tolerancia a todas las acciones de las benzodiazepinas puede desarrollarse, aunque a tasas variables y en diferentes grados. La tolerancia a los efectos hipnóticos tiende a desarrollarse rápidamente, lo que puede ser beneficioso en la ansiolisis diurna, pero dificulta el tratamiento a largo plazo del insomnio.17 Los pacientes suelen notar alivio de insomnio inicialmente, seguido de una pérdida gradual de eficacia.18 La tolerancia al efecto ansiolítico parece desarrollarse más lentamente que la tolerancia a los efectos hipnóticos, pero existe poca evidencia que indique que las benzodiazepinas conserven su eficacia después de cuatro a seis meses de uso regular.19, 20. La terapia con benzodiazepinas a menudo se continúa para suprimir los estados de abstinencia, que generalmente imitan los síntomas de ansiedad. El aumento de la dosis a menudo mantiene el ciclo de tolerancia y dependencia, y los pacientes pueden tener dificultades para interrumpir la terapia con medicamentos.
Dependencia
El tratamiento con benzodiazepinas puede dar lugar a dependencia fisiológica y psicológica según la dosis del fármaco, la duración del tratamiento y la potencia.1 Por lo tanto, la dependencia se desarrollará más pronto (como en uno o dos meses) en un paciente que toma una dosis alta de un agente de alta potencia como el alprazolam que en un paciente que recibe una dosis relativamente baja de un fármaco de acción prolongada. Agente de baja potencia como el clordiazepóxido. Como resultado de la dependencia fisiológica, los síntomas de abstinencia emergen con una reducción rápida de la dosis o una interrupción brusca del medicamento.
Psicológicamente, el uso a largo plazo de benzodiazepinas puede conducir a un exceso de confianza en la necesidad del agente, la pérdida de confianza en sí mismo y los diversos grados de comportamiento de búsqueda de drogas.8 Los pacientes pueden mostrarse reacios a suspender el medicamento debido a temores fuera de lugar o ansiedad anticipada. Algunos pacientes combinan alcohol con benzodiacepinas cuando no pueden adquirir los efectos deseados o «necesarios».
Síntoma de abstinencia a corto plazo
Los efectos de abstinencia de las dosis terapéuticas de las benzodiazepinas son principalmente síntomas de ansiedad.1,21 Además, son comunes la inestabilidad autónoma (es decir, el aumento de la frecuencia cardíaca y el nivel de presión arterial, temblores, diaforesis), el insomnio y la hipersensibilidad sensorial. Los síntomas más graves de abstinencia aguda son las convulsiones y el delirium tremens, que con mayor frecuencia ocurren con la interrupción brusca. El marco de tiempo para la aparición de síntomas agudos de abstinencia corresponde a la vida media del agente particular que se está utilizando. Se cree que algunos elementos de abstinencia ocurren en la mayoría de los pacientes que han tomado dosis terapéuticas de benzodiazepinas durante más de unos pocos meses, aunque la gravedad de los síntomas de abstinencia generalmente depende de la cantidad de la dosis original, la velocidad a la que la dosis es cónico, la selección de pacientes y la definición de síntomas de abstinencia.1,18
Abstinencia proactiva
Los psicólogos que están familiarizados con la adicción a las benzodiazepinas han observado un síndrome de abstinencia prolongada.22 Los síntomas incluyen ansiedad prolongada (por varios meses), depresión e insomnio. Además, pueden aparecer síntomas físicos relacionados con efectos gastrointestinales, neurológicos y musculoesqueléticos. Este fenómeno de abstinencia puede desarrollarse a pesar de una reducción lenta, juiciosa y juiciosa de la dosis y se cree que es el resultado de una neuroadaptación crónica.
Efectos en pacientes ancianos
Entre los ancianos, se puede amplificar el riesgo de interacciones farmacológicas, ralentización psicomotora, disfunción cognitiva y desinhibición paradójica. El uso de benzodiazepinas en los ancianos se asocia con una mayor tasa de caídas que causan fracturas de cadera y fémur y una mayor probabilidad de accidentes automovilísticos.23,24 El deterioro cognitivo es común, aunque el deterioro de la memoria puede ser reversible cuando se suspenden las benzodiazepinas.25
El deterioro cognitivo asociado con los procesos de envejecimiento normales y la demencia puede empeorar debido a los efectos secundarios de las benzodiazepinas. Los mecanismos de supresión cortical se pueden alterar en los ancianos y los comportamientos desinhibidos pueden aumentar con el uso de benzodiazepinas. Con menos reserva cognitiva y social en el paciente anciano, los síntomas de abstinencia a corto y largo plazo y otros efectos secundarios de las benzodiazepinas pueden llevar al paciente a visitar o llamar al médico con frecuencia. El médico puede sentirse «atrapado» en la discusión contra el uso de benzodiazepinas y la prescripción de benzodiazepinas a pacientes de edad avanzada. En un estudio,26, este impasse se rompió al remitir a los pacientes ancianos a la desintoxicación hospitalaria, lo que resultó en una disminución dramática en las visitas anuales al médico.
Abuso de benzodiazepinas
Las benzodiazepinas rara vez son la droga preferida o la única de abuso. Se estima que el 80 por ciento del abuso de benzodiazepinas es parte del abuso de drogas múltiples, más comúnmente con opioides.27 Un estudio de dos años sobre el resultado del tratamiento realizado por el Instituto Nacional sobre el Abuso de Drogas 28 encontró que el 15 por ciento de los usuarios de heroína también usaban benzodiazepinas diariamente durante más de un año, y el 73 por ciento usaba benzodiazepinas con más frecuencia que semanalmente. Los estudios indican que desde el 5 por ciento hasta el 90 por ciento de los usuarios de metadona también son usuarios habituales de benzodiazepinas. El abuso de altas dosis de benzodiazepinas prevalece especialmente en pacientes que toman metadona. 29
Los estudios indican que del 3 al 41 por ciento de las personas alcohólicas informan que abusaron de las benzodiazepinas en algún momento, a menudo para modular la intoxicación o los efectos de abstinencia.4 El alcohólico contemporáneo es usualmente un usuario de múltiples drogas. Hasta el 80 por ciento de los alcohólicos menores de 30 años han sido adictos o usan al menos otra droga.27
Las recetas médicas constituyen la principal fuente de suministro para las personas que abusan de las benzodiazepinas. Las recetas también pueden tener un valor en la calle, lo que alienta el desvío a fuentes ilícitas. Las benzodiazepinas tienen múltiples usos para los adictos a las drogas múltiples: se usan para mejorar los efectos de euforia de los opioides (como para «aumentar» las dosis de metadona), para aliviar los síndromes de abstinencia o abstinencia (como los «arreglos» de heroína), para atenuar los niveles altos de cocaína. aumentar el alcohol sinérgicamente y modular los estados de abstinencia.
Como posibles drogas de abuso, las benzodiazepinas de acción corta parecen ser preferidas entre los adictos debido a la rapidez de su acción.30 En general, las sustancias que alteran el estado de ánimo son más altamente reforzantes en pacientes con dependencia química si el agente tiene un inicio de acción rápido, una alta potencia, una breve duración de la acción, alta pureza y solubilidad en agua (para uso intravenoso) o alta volatilidad (Capacidad de vaporizar si se fuma).31 Los datos sugieren que las benzodiazepinas altamente lipofílicas (por ejemplo, las que cruzan la barrera hematoencefálica más rápidamente), como el diazepam, y los agentes con una vida media corta y alta potencia, como el lorazepam o el alprazolam, son las benzodiazepinas más reforzantes y, por lo tanto, los más propensos a estar asociados con el abuso.30
Clonazepam es una benzodiazepina de alta potencia con una larga vida media. Se prescribe ampliamente para una variedad de condiciones psiquiátricas y neurológicas. Aunque el clonazepam se percibe como «seguro», los especialistas en adicción a la medicina han descubierto que también se abusa frecuentemente como una droga callejera. Por otro lado, el oxazepam (Serax), el clorazepato (Tranxene) y el clordiazepóxido parecen tener efectos de refuerzo más bajos que otras benzodiazepinas.
En comparación con las formulaciones genéricas, los medicamentos recetados de marca comercial pueden valer el doble por tableta cuando se venden en la calle porque son fácilmente reconocibles como «reales» cuando se comparan con las fotografías de tabletas en la Referencia de escritorio de los médicos .31 Las píldoras genéricas a menudo son irreconocibles y, por lo tanto, valen menos cuando se desvían para la venta callejera. En muchas ciudades de los EE. UU., El valor en la calle de Xanax o Klonopin puede ser de $ 5 a $ 10 por píldora, según la dosis.
Alternativas a la benzodiazepina
Los problemas con la dependencia, la tolerancia, el retiro, el rebote y el abuso de las benzodiazepinas limitan su uso para el tratamiento a largo plazo de los trastornos de ansiedad en pacientes con adicción al alcohol o drogas. Un creciente cuerpo de literatura ahora apoya la eficacia ansiolítica de muchos otros agentes (Tabla 3). Se ha demostrado que los antidepresivos, anticonvulsivos, buspirona (Buspar), ciertos agentes antihipertensivos y neurolépticos más nuevos son eficaces en subconjuntos de pacientes con ansiedad.32
Tabla 3
Eficacia de los agentes farmacológicos en el tratamiento de los trastornos de ansiedad
TRASTORNO
BZS
ISRS
ATC
ACVS*
BU
AN†
AHTS‡
Ansiedad aguda
++
–
–
–
–
+
+
Trastorno de ansiedad generalizada
++
+
++
±
++
–
–
Trastorno de pánico
++
++
++
+
–
–
–
Fobia social
+
++
+
–
+
–
–
Trastorno de estrés postraumático
±
+
+
+
+
+
–
Desorden obsesivo compulsivo
–
++
+
–
+
±
–
BZs = benzodiazepinas; ISRS = inhibidores selectivos de la recaptación de serotonina; ATC = antidepresivos tricíclicos; ACVs = anticonvulsivos; Bu = Buspirona (Buspar); ANs = neurolépticos atípicos; HTA = antihipertensivos . ++ = eficacia probada en numerosos ensayos controlados; + = eficacia informada en ensayos abiertos o en pacientes con depresión comórbida; ± = eficacia equívoca, informes anecdóticos o uso complementario; – = No hay buena evidencia clínica de eficacia . *: Los anticonvulsivos incluyen ácido valproico (Depakene) y gabapentina (Neurontin) . †: los neurolépticos atípicos incluyen risperidona (Risperdal), olanzapina (Zyprexa) y quetiapina (Seroquel) . ‡: los antihipertensivos incluyen bloqueadores beta y clonidina (Catapres) .
La mayoría de los especialistas en medicina de adicción creen que las benzodiazepinas están relativamente contraindicadas en pacientes con problemas actuales de abuso de alcohol o drogas y en pacientes en recuperación. Para elegir una alternativa adecuada a una benzodiazepina, los médicos deben poder delinear qué subtipo de trastorno de ansiedad existe en un paciente en particular. Se debe alentar a los pacientes a comprender que el inicio de la acción de los antidepresivos, la buspirona y los anticonvulsivos no es tan inmediato como el de las benzodiazepinas. La terapia puede requerir paciencia y, debido a los efectos secundarios, puede requerirse inicialmente una dosis baja.
Insomnio
El insomnio es una secuela común de numerosas afecciones médicas y psiquiátricas, y a menudo se asocia con trastornos por uso de sustancias, abstinencia temprana o abstinencia prolongada. El manejo del insomnio incluye atención a las técnicas de higiene del sueño, como mantener un ciclo regular de sueño y vigilia, evitar las siestas durante el día, evitar la cafeína o las comidas pesadas por la noche, y realizar ejercicios suaves o utilizar otras técnicas de relajación.
Las farmacoterapias que no son de benzodiazepina para el tratamiento del insomnio incluyen el antidepresivo sedante trazodona (Desyrel), los antidepresivos tricíclicos terciarios como la amitriptilina (Elavil) y la doxepina (Sinequan), y los agentes antidepresivos más nuevos como la nefazodona y la mirtazacina (Presencia). 33
Zolpidem (Ambien), una imidazopiridina, es un agente hipnótico con una estructura química no relacionada con las benzodiazepinas.34 A diferencia de las benzodiazepinas, el zolpidem no interfiere con las etapas 3 y 4 del sueño, ni disminuye el sueño con movimientos rápidos de los ojos (REM). Los síntomas de tolerancia y abstinencia no aparecen tan fácilmente con este agente como con las benzodiazepinas. Sin embargo, el zolpidem se clasifica como una sustancia controlada en la lista IV (como las benzodiazepinas) y se han observado efectos sinérgicos con las benzodiazepinas y el alcohol. También se han reportado problemas con sueños vívidos, pesadillas e insomnio de rebote.34
Comentario Final
Aunque las benzodiazepinas son efectivas en una amplia gama de afecciones médicas y psiquiátricas, se debe tener precaución con su uso, particularmente cuando estos agentes se prescriben a pacientes con un historial activo o remoto de abuso de sustancias o adicción. Su mayor activo es también su mayor responsabilidad: las drogas que funcionan de inmediato tienden a ser adictivas. En comparación con las benzodiazepinas, los antidepresivos tienen un inicio de acción más prolongado, pero son los mejores agentes para el tratamiento a largo plazo de los trastornos de ansiedad. Los anticonvulsivos, los antipsicóticos, los antihipertensivos y la buspirona también son efectivos, pero tienen un inicio de acción intermedio.
El juicio clínico se basa en una evaluación de los riesgos frente a los beneficios de la terapia. Dicho enfoque podría tener en cuenta si el abuso de sustancias es activo o remoto, si otros miembros de la familia u otros profesionales de la salud están involucrados activamente en la atención del paciente y qué tan bien el médico conoce al paciente. Los médicos también deben buscar libremente la consulta de especialistas como psiquiatras y especialistas en medicina de adicciones. La educación, la consulta y la documentación no solo mejoran el nivel de atención clínica, sino que también proporcionan la gestión de riesgos necesaria y la protección de responsabilidad legal.
Referencias
Salzman C, for Task Force on Benzodiazepine Dependency, American Psychiatric Association. Benzodiazepine dependence, toxicity, and abuse: a task force report of the American Psychiatric Association. Washington, D.C.: American Psychiatric Association, 1990.
American Druggist. Top 200 drugs of 1995. New York, N.Y.: Hearst Corp, 1996:18–26.
Hollister LE, Muller-Oerlinghausen B, Rickels K, Shader RI. Clinical uses of benzodiazepines. J Clin Psychopharmacol. 1993;13(suppl 1):1S–169S.
Ciraulo DA, Sands BF, Shader RI. Critical review of liability for benzodiazepine abuse among alcoholics. Am J Psychiatry. 1988;145:1501–6.
Busto UE, Romach MK, Sellers EM. Multiple drug use and psychiatric comorbidity in patients admitted to the hospital with severe benzodiazepine dependence. J Clin Psychopharmacol. 1996;16:51–7.
Arana GW, Hyman SE. Handbook of psychiatric drug therapy. 2d ed. Boston: Little, Brown, 1991: 128–61.
Miller LG. Chronic benzodiazepine administration: from the patient to the gene. J Clin Pharmacol. 1991;31:492–5.
Ashton H. Toxicity and adverse consequences of benzodiazepine use. Psychiatric Annals. 1995;25:158–65.
Janicak PG, Davis JM, Preskhorn SH, Ayd FJ. Principles and practice of psychopharmacotherapy. Baltimore: Williams & Wilkins 1993:287.
Geene DS, Dockens RC, Salazar DE, et al. Coadministration of nefazadone and benzodiazepines. I. pharmacokinetic assessment [Abstract]. Clin Pharmacol Ther. 1994;55:141.
Lader M. Long-term benzodiazepine use and psychological functioning. In: Freeman HL, Rue Y, eds. The benzodiazepines in current clinical practice. International congress and symposium series: proceedings of a symposium sponsored by Wyeth Laboratories. London: Royal Society of Medicine Services, 1987:55–69.
Barbone F, McMahon AD, Davey PG, Morris AD, Reid IC, McDevitt DC, et al. Association of road-traffic accidents with benzodiazepine use. Lancet. 1998;352:1331–6.
Curran V. Memory functions, alertness, and mood of long-term benzodiazepine users: a preliminary investigation of the effects of normal daily dose. J Psychopharmacol. 1992;6:69–75.
van der Bijl P, Roelofse JA. Disinhibitory reactions to benzodiazepines: a review. J Oral Maxillofac Surg. 1991;49:519–23.
Smith BD, Salzman C. Do benzodiazepines cause depression? Hosp Comm Psychiatry. 1991;42:1101–2.
Bergman U, Rosa FW, Baum C, Wiholm BE, Faich GA. Effects of exposure to benzodiazepines during fetal life. Lancet. 1992;340:694–7.
Kales A, Scharf MB, Kales JD. Rebound insomnia: a new clinical syndrome. Science. 1978;201:1039–41.
Schneider-Helmert D. Why low-dose benzodiazepine-dependent insomniacs can’t escape their sleeping pills. Acta Psychiatr Scand. 1988;78:706–11.
Mellinger GD, Balter MB, Uhlenhuth EH. Prevalence and correlates of the long-term use of anxiolytics. JAMA. 1984;251:375–9.
Marriott S, Tyrer P. Benzodiazepine dependence. Avoidance and withdrawal. Drug Saf. 1993;9:93–103.
Busto U, Sellars EM, Naranjo CA, Cappell H, Sanchez-Craig M, Sykora K. Withdrawal reaction after long-term therapeutic use of benzodiazepines. N Engl J Med. 1986;315:854–9.
Ashton H. Protracted withdrawal syndromes from benzodiazepines. J Subst Abuse Treat. 1991;8:19–28.
Herings RM, Stricker BH, de Boer A, Bakker A, Sturmans F. Benzodiazepines and the risk of falling leading to femur fractures. Dosage more important than elimination half-life. Arch Intern Med. 1995;55:1801–7.
Hemmelgarn B, Suissa S, Huang A, Boivin IF, Pinard G. Benzodiazepine use and the risk of motor vehicle crash in the elderly. JAMA. 1997;278:27–31.
Salzman C, Fisher J, Nobelk Glassman R, Wolfson A, Kelly M. Cognitive improvement following benzodiazepine discontinuation in elderly nursing home residents. Int J Geriatr Psychiatry. 1992;7:89–93.
Burke KC, Meek WJ, Krych R, Nisbett R, et al. Medical services use by patients before and after detoxification from benzodiazepine dependence. Psychiatric Services. 1995;46(2):157–60.
Gold MS, Miller NS, Stennie K, Populla-Vardi C. Epidemology of benzodiazepine use and dependence. Psychiatric Annals. 1995;25:*146–8.
Dumont RL. Abuse of benzodiazepines—the problems and the solutions. A report of a Committee of the Institute for Behavior and Health, Inc. Am J Drug Alcohol Abuse. 1988;14(suppl 1):1–69.
Iguchi MY, Griffiths RR, Bickel WK, et al. Relative abuse liability of benzodiazepines in methadonemaintained populations in three cities. In: Harris LS, ed. Problems of drug dependence, 1988: proceedings of the 50th Annual Scientific Meeting, the Committee on Problems of Drug Dependence, Inc. Rockville, Md.: U.S. Department of Health and Human Services, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration,National Institute on Drug Abuse, Office of Science, 1989. DHHS publication no. (ADM) 89-1605.
Roache JD, Meisch RA. Findings from self-administration research on the addiction potential of benzodiazepines. Psychiatric Annals. 1995;25(3):153–7.
Parran TV. Prescription drug abuse. A question of balance. Med Clin North Am. 1997;81:967–78.
Longo LP. Non-benzodiazepine pharmacotherapy of anxiety and panic in substance abusing patients. Psychiatric Annals. 1998;28(3):142–53.
Longo LP, Johnson B. Treatment of insomnia in substance abusing patients. Psychiatric Annals. 1998;28(3):154–9.
Zolpidem for insomnia. Med Lett Drugs Ther. 1993;35:35–6.
Referencias de autores:
LANCE P. LONGO, M.D., University of Wisconsin Medical School, Milwaukee, WisconsinM.D.
BRIAN JOHNSON, Harvard Medical School, Boston, Massachusetts
La afirmación de que la adicción es una enfermedad cerebral es casi universalmente aceptada entre los científicos que trabajan en la adicción. La atracción de la afirmación se basa en dos motivos: el hecho de que la adicción parece estar caracterizada por una disfunción en vías neuronales específicas y el hecho de que la reclamación parece ser la respuesta compasiva a las personas que sufren. Argumento que la disfunción neural no es suficiente para la enfermedad: algo es una enfermedad cerebral solo cuando la disfunción neural es suficiente para el deterioro. Afirmo que la disfunción neural que es característica de la adicción no es suficiente para el deterioro, porque las personas que la padecen están dañadas, lo suficiente como para considerarse enfermas y solo se les dan ciertas características de su contexto. Por lo tanto, la adicción no es una enfermedad cerebral (aunque a menudo es una enfermedad, y siempre puede implicar disfunción cerebral). Sostengo que aceptar que la adicción no es una enfermedad cerebral no implica una actitud moralizadora hacia las personas que sufren como resultado de la adicción; En todo caso, permite una respuesta más compasiva y más efectiva a la adicción.
Los neurocientíficos y otros científicos que participan en el estudio de la adicción ven correctamente su trabajo no solo como una ciencia objetiva sino también como un proyecto compasivo. Su objetivo no solo es dilucidar las causas neuropsicológicas y los correlatos de la adicción, sino también proporcionar un conocimiento que pueda aplicarse en el tratamiento de las personas que sufren. Incluso aquellos cuyo trabajo está muy alejado del aspecto clínico, aquellos que trabajan en modelos animales de adicción, por ejemplo, consideran que sus hallazgos y los de sus compañeros tienen implicaciones importantes sobre cómo deberíamos responder a los adictos. La elucidación de los fundamentos neurales de la adicción muestra que la adicción es una enfermedad que debe tratarse, no algo por lo que se puede culpar a los adictos. Como Leshner (1997) ha dicho, la adicción es una enfermedad cerebral, y es importante.
En este artículo, argumentaré que el eslogan es, en el mejor de los casos, engañoso. La adicción no se entiende mejor como una enfermedad cerebral, aunque ciertamente implica una disfunción neuropsicológica patológica. La adicción es un trastorno de una persona, incrustado en un contexto social. Los neurocientíficos y sus aliados han confundido algunas condiciones necesarias del trastorno con el propio trastorno [1]. A pesar de esta afirmación, hay, sin embargo, un caso sólido para decir que la adicción es a menudo una enfermedad. Restaurar a los adictos a sus contextos sociales no requiere que aceptemos la visión de la adicción a la que se oponen los neurocientíficos, el modelo moral. Más bien, podemos situar al adicto en un contexto social, e incluso reconocer que los juicios sobre el desorden son parcialmente normativos, sin abandonar un marco completamente naturalista.
Los neurocientíficos adoptan el modelo de adicción a la enfermedad cerebral por una razón obvia: porque han hecho grandes progresos en el esclarecimiento de los mecanismos neuronales y las neuroadaptaciones que están correlacionadas con, y sin duda, causalmente involucradas en la adicción. Los neurocientíficos han identificado un rango de tales cambios, que incluyen (pero no se limitan a) la depresión a largo plazo de los circuitos de recompensa y el aumento de la actividad en los circuitos antibalas (Koob y Le Moal, 1997, 2008); alteraciones en el sistema de dopamina del cerebro medio (Volkow y Li, 2004); y en regiones frontales involucradas en la inhibición de impulsos (Goldstein y Volkow, 2002). Sin embargo, hay cambios neuronales asociados y causalmente involucrados en todos los comportamientos Establecer que esto es cierto con respecto a la adicción, por lo tanto, no establece que se trata de una enfermedad cerebral.
¿Qué se necesitaría para demostrar que la adicción es una enfermedad cerebral? Los detalles de los correlatos neurales de la adicción son importantes: la adicción es una enfermedad cerebral si estos correlatos son patológicos y si esa patología es suficiente para que la persona tenga una enfermedad, en casi cualquier entorno accesible (más adelante hablaré sobre esta condición). Argumentaré que, aunque hay razones para decir que los correlatos de la adicción son patológicos, estos correlatos no son suficientes para que la persona tenga una enfermedad en algunos entornos accesibles. Independientemente de si los correlatos son en sí mismos patológicos, la persona tiene una enfermedad solo en la medida en que su funcionamiento como agente está deteriorado, y en muchos entornos los correlatos de la adicción no son suficientes para el deterioro. Además, sugeriré, los juicios sobre deterioro son juicios normativos, donde las normas en cuestión no son normas de funcionamiento cerebral. Sin embargo, dado que un juicio normativo no tiene por qué ser un juicio no naturalista – las protestas de muchos, al contrario – aceptar que el juicio de que la adicción es una enfermedad es parcialmente normativo, no requiere aceptar el modelo moral de adicción, ya que ese modelo es estándar concebido.
¿Son los correlatos neurales de la adicción patológicos?
Si el juicio de que la adicción es una enfermedad no tiene vergüenza de ser normativo, y las normas en cuestión no son normas de la función cerebral, entonces la adicción no es una enfermedad cerebral. La adicción es una enfermedad cerebral solo si las desviaciones patológicas de las normas de la función cerebral son (en casi cualquier entorno accesible) suficientes para estar alteradas. Sin embargo, el hecho de que la adicción sea causada por una disfunción patológica del cerebro no es tan obvio como parece a muchos científicos. Existen relatos científicos sobre la adicción según los cuales no implica ninguna patología cerebral. Sobre las teorías que tengo en mente, explicar la adicción nos obliga a postular mecanismos cerebrales no patológicos.
Considere una cuenta de la adicción no coincidente (Durrant et al., 2009). Las cuentas de desajuste se centran en un desajuste entre nuestras capacidades y disposiciones evolucionadas, por un lado, y el entorno en el que muchas personas se encuentran hoy, por el otro. Este tipo de hipótesis parece una explicación plausible (parcial) de la epidemia de obesidad actual. A grandes rasgos, la idea es la siguiente: en el entorno de adaptabilidad evolutiva, las calorías eran relativamente escasas. Por lo tanto, fue adaptativo desarrollar disposiciones para consumir la mayor cantidad posible de alimentos altos en calorías (dulces o grasos) cuando estuvieran disponibles, dado que estos alimentos no se podían almacenar por mucho tiempo. Hoy, sin embargo, los alimentos grasos y azucarados están abundantemente disponibles, pero estamos dispuestos a consumirlos más allá de las necesidades inmediatas y a corto plazo. Debido a que no necesitamos ejercer el autocontrol en el EEE con respecto a estos alimentos, sino todo lo contrario, dependemos de mecanismos limitados, de arriba a abajo y de dominio general para que el autocontrol resista el consumo excesivo, y estos mecanismos son: relativamente fácil de eludir o agotado. De ahí la epidemia de obesidad. Al igual que en ausencia de un mecanismo de razonamiento específico de dominio para condicionales, estamos obligados a utilizar el razonamiento de dominio general para su evaluación y predeciblemente mal (Cosmides y Tooby,1992), en ausencia de mecanismos de autocontrol específicos de la sustancia, somos devueltos a nuestros mecanismos de autocontrol de dominio general, y el consumo excesivo es un resultado probable.
Ha habido intentos de desarrollar cuentas desajustadas de enfermedades mentales. Por ejemplo, Murphy y Stich (2000) han planteado la hipótesis de que a veces la depresión puede resultar de un detector de estado relativo extremadamente sensible (pero no patológicamente). Su propuesta se basa en Nesse y Williams (1995) y sugieren que la depresión puede ser una respuesta adaptativa a una caída o un fracaso para obtener el estado. Tales fallas desencadenan una disposición que enfoca al individuo hacia adentro (de ahí la rumia característica de la depresión) por lo que los alienta a identificar estrategias sociales infructuosas y a desarrollar nuevas, y hace que se retiren del contacto social que podría desencadenar la agresión por parte de los miembros dominantes del grupo. Murphy y Stich proponen que cuando el detector de estatus social relevante se dirige hacia el final más sensible de la distribución normal, se activará continuamente en las sociedades contemporáneas en las que tenemos oportunidades constantes de compararnos con individuos de alto estatus (celebridades, magnates, atletas, y así sucesivamente) en todo el mundo. El resultado es el desencadenamiento de la depresión.
El consumo de sustancias que alteran la mente se remonta a miles de años en la historia de la humanidad, y muy plausiblemente comenzó en la prehistoria. Los registros de uso de opio se remontan a casi 6000 años (Booth, 1996); La elaboración de cerveza se remonta aún más. Sin embargo, es poco probable que alguien haya tenido acceso a una cantidad suficiente de dichas sustancias, durante un período de tiempo suficientemente prolongado, para que estas sustancias generen serios problemas hasta hace muy poco tiempo, cuando la agricultura se generalizó y los humanos se volvieron sedentarios (Durrant et al., 2009); de hecho, es muy probable que el autocontrol con respecto a ellos primero fuera requerido solo con el crecimiento de las ciudades. En estas condiciones, no hubo presión selectiva para que los seres humanos desarrollen un mecanismo específico de autocontrol con respecto a estas sustancias.
Hoy en día, estas sustancias están abundantemente disponibles, al menos para algunas personas y en algunos lugares (alcohol y tabaco para la mayoría de las personas en países desarrollados; otras sustancias para subgrupos particulares; la adicción se correlaciona con un estatus socioeconómico más bajo y las drogas adictivas tienden a ser más abundantes en las zonas bajas del SES). Debido a que carecemos de un mecanismo de autocontrol específico de la sustancia, se nos recurre a recursos de autocontrol de dominio general, y estos recursos se agotan fácilmente (Baumeister y Vohs, 2007).
Sin embargo, hay una diferencia sobresaliente entre los alimentos ricos en calorías y las drogas de adicción: es plausible que la evolución nos deje con disposiciones para perseguir la primera, pero no hay evidencia de que tengamos disposiciones innatas para perseguir la segunda ( ritmo de Sullivan y Hagen, 2002). Sin embargo, el gusto por las drogas puede ser adquirido; no hay razón por la cual una disposición adquirida deba ser más débil que una innata. Además, incluso si la disposición a usar drogas es menos motivadora, o motiva menos acciones individuales, que la disposición innata a consumir alimentos, los daños asociados con las drogas se acumulan mucho más rápidamente, por lo que una teoría que postule una disposición más débil puede explicar los efectos observados.
La teoría del desajuste parece recorrer un largo camino para explicar los problemas que muchas personas tienen con las sustancias adictivas. También ayuda a explicar por qué los factores ambientales que limitan el acceso a estas sustancias son muy protectores. Pero la cuenta del desajuste claramente no implica que la adicción sea una enfermedad cerebral. La razón es simple: si la cuenta de discordancia explica (o, más plausiblemente, tiene un papel importante en la explicación) de la adicción, entonces implica que la adicción se explica por los mecanismos cerebrales que funcionan como están diseñados (Murphy y Stich, 2000). En la medida en que la cuenta de discordancia explica por qué a los adictos les resulta difícil resistirse a las drogas adictivas, implica que no padecen en absoluto una patología cerebral.
Si queremos mostrar que la adicción es una enfermedad cerebral, deberemos demostrar que la patología subyacente es una patología del cerebro. Necesitamos mostrar que el cerebro es disfuncional, de la misma manera que los científicos médicos establecen que un órgano está enfermo al demostrar que es disfuncional. El ejemplo canónico en la ciencia médica es la enfermedad cardíaca: la enfermedad cardíaca se considera una enfermedad porque amenaza con interferir con la función del corazón. El papel funcional del corazón es bombear sangre; Porque la enfermedad cardíaca interfiere con ese papel, es una enfermedad.
Las cuentas de disfunción vienen en dos variedades, correspondientes a los dos análisis filosóficos de la función que compiten entre sí. En una cuenta seleccionista, expuesto más influyente por Millikan (1984), una disfunción se produce cuando algo falla en desempeñar el papel para el que fue seleccionado en la historia evolutiva del organismo. En la cuenta sistémica, desarrollada por Cummins (1975), no es el papel que algo jugó en la historia evolutiva lo que le da su función; más bien, es el papel que desempeña (o sus homólogos) en un sistema. No tengo la intención de tratar de resolver el debate entre estas cuentas. Más bien, me centraré en lo que las cuentas tienen en común, argumentando que ninguna de las dos implica que la adicción es una enfermedad cerebral.
Hay teorías plausibles que explican (en parte) la adicción e implican que el cerebro del adicto es disfuncional. Supongamos que algún tipo de cuenta dopaminérgica de la adicción es correcta; Supongamos, es decir, que la adicción implica una patología en el sistema de dopamina del cerebro medio. La opinión generalizada es que el sistema dopaminérgico del cerebro medio es un sistema de valoración: tiene la función de señalar el valor de un recurso para el organismo y motivar al organismo hacia el consumo de ese recurso. Esta visión se deriva, en gran parte, de un trabajo importante de Schultz et al. En varios experimentos, Schultz et al. registró la actividad de las neuronas dopaminérgicas del cerebro medio en monos que realizan diversas tareas que fueron recompensadas con agua o jugo. En un experimento, los monos aprendieron que recibirían una recompensa si presionaban una palanca en particular,1992). Durante la fase de aprendizaje, las neuronas respondieron enérgicamente a la entrega de la recompensa, pero una vez que se aprendió la tarea y la asociación entre la señal y la disponibilidad de jugo, las neuronas respondieron cuando se dio la señal, pero no cuando se entregó la recompensa. De manera similar, las neuronas de dopamina en monos responden inicialmente a la entrega de una recompensa predicha por una señal visual, pero a medida que se aprende la asociación entre la señal y la recompensa, la respuesta a la recompensa disminuye mientras que la respuesta a la señal predice la recompensa Incrementos (Sutton y Barto, 1998).
Sobre la base de este tipo de evidencia, muchos investigadores han llegado a creer que el sistema mesolímbico es un sistema de predicción de recompensa (Montague et al., 1996; Schultz et al., 1997). Nos permite aprender el valor de una recompensa y la relación entre las señales ambientales y las recompensas. Esta función es obviamente adaptativa, ya que desempeña un papel crucial para guiar y motivar al organismo en la búsqueda de recompensas, donde las “recompensas” son bienes necesarios para la supervivencia y la reproducción.
Sin embargo, la adicción parece implicar una desregulación en este mismo sistema de dopamina del cerebro medio. Casi todas las drogas adictivas aumentan la actividad dopaminérgica. La anfetamina, la nicotina, el cannabis, la cocaína y el alcohol estimulan la liberación de dopamina o disminuyen la recaptación de dopamina. Con ello aumentan la dopamina en el núcleo accumbens. Los opioides aumentan la dopamina indirectamente, al influir en las neuronas que alteran la dopamina accumbal (Carter y Hall, 2012). La cafeína también aumenta la dopamina extracelular en el núcleo accumbens (Solinas et al., 2002). La forma en que las drogas adictivas (y, de una manera muy diferente, el juego; ver Ross et al., 2008Se considera que el aumento de la señal de la dopamina es fundamental para explicar cómo se desarrolla la adicción y por qué es una afección crónica recurrente. Para muchos expertos en adicciones, la adicción es una patología del sistema dopaminérgico. En la metáfora común, las drogas adictivas «secuestran» este sistema. Es decir, la adicción implica crucialmente un sistema mesolímbico disfuncional.
La hipótesis de la predicción de la recompensa parece explicar la adicción entendiéndola como una patología del aprendizaje por refuerzo. Cuando el sistema está funcionando como debería, la activación dopaminérgica se atenúa en respuesta a la recompensa esperada. La respuesta de la dopamina aumenta cuando el mundo es mejor de lo esperado; cuando se entrega una recompensa esperada, el mundo es exactamente como se esperaba y no debería haber respuesta de dopamina. Si los medicamentos funcionaran como recompensas naturales, podríamos esperar que desencadenen una respuesta inicial de dopamina al consumo, pero se atenúa esta respuesta a medida que se repite el consumo. Al mismo tiempo, deberíamos esperar un aumento en la respuesta de la dopamina a los factores predictivos de la disponibilidad de medicamentos. En cambio, lo que encontramos es la respuesta de la dopamina a los factores predictivos de la disponibilidad de medicamentos y- debido a que las drogas de la adicción aumentan la respuesta de la dopamina por su acción química – la actividad dopaminérgica continua en el consumo también. En efecto, el sistema dopaminérgico responde a los medicamentos con la señal de que el consumo es mejor de lo esperado. Lo hace cada vez que se consume la droga. El adicto no puede aprender el valor de recompensa de la droga, porque el sistema para el aprendizaje del valor de recompensa es disfuncional. En cada ocasión en que se consume el medicamento, el sistema dopaminérgico informa que el medicamento es más gratificante de lo esperado. El resultado es el aprendizaje patológico; El sistema trata la droga como de valor cada vez mayor.
Cabe señalar que hay rivales en la interpretación de la predicción de la recompensa de la actividad dopaminérgica mesolímbica. Berridge, 2007sugiere que el papel de la dopamina es incentivar la prominencia, no el aprendizaje. Berridge señala que aprender sobre la relación entre un estímulo y una recompensa puede ocurrir sin la dopamina. En ratones genéticamente diseñados para ser incapaces de sintetizar dopamina, el aprendizaje normal parece ocurrir. También ocurre en ratones que prácticamente no tienen dopamina mesolímbica debido a lesiones neuroquímicas. Además, la activación en el pálido ventral, aguas abajo del sistema de dopamina mesolímbico, es más fuerte en respuesta a un segundo, redundante, predictor de recompensa que en respuesta al primero. Dado que el segundo predictor no agrega información nueva, deberíamos esperar una respuesta más pequeña al segundo predictor que al primero si el sistema de dopamina era en sí mismo un sistema de predicción de recompensa.
Para Berridge (2007); Holton y Berridge, de próxima aparición, la adicción es una patología de prominencia de incentivo y no recompensa predicción. No implica aprendizaje patológico; más bien implica un “deseo” patológico. Podemos dejar esta disputa a un lado. Para nuestros propósitos, lo que importa es lo que los investigadores acuerdan: que la disfunción en el sistema dopaminérgico es, o refleja, una disfunción de un sistema que evolucionó para jugar (o normalmente juega) un papel específico en el comportamiento, ya sea Aprendizaje o incentivo de la saliencia. En cualquiera de las dos historias, podríamos entender el sistema que está mal, ya sea el sistema de dopamina del cerebro medio o algo corriente arriba de ese sistema, como algo que representa los bienes en el mundo externo, como lo valiosos que son para el organismo. En cualquier historia, La adicción causa una tergiversación y por eso es una patología. Es una patología porque el sistema fue seleccionado para desempeñar, o en realidad desempeña, un papel particular en la psicología de los animales como nosotros, pero ya no juega ese papel en los adictos, al menos en respuesta a las drogas y las señales que predicen la disponibilidad de drogas.
Ambas explicaciones de la disfunción, junto con hipótesis plausibles sobre los correlatos neurales de la adicción, implican que la adicción implica una disfunción neuropsicológica. Sin embargo, ninguna explicación implica que la adicción es una enfermedad cerebral. Anteriormente afirmé que la adicción es una enfermedad cerebral solo si se satisfacen dos condiciones: sus correlatos neurales son patológicos y que la patología es suficiente para que la persona sufra una enfermedad en casi cualquier entorno accesible. Esta segunda condición es necesaria para descartar las condiciones en las que la respuesta adecuada al sufrimiento es alterar el entorno y no «tratar» a la persona. Considera la homosexualidad. Sigue siendo una pregunta abierta si los correlatos neuronales de la homosexualidad son adaptativos. De Wilson (1975) la conjetura, por ejemplo, según la cual la homosexualidad existe hoy en día debido a la selección dependiente de la frecuencia que mantiene los genes relevantes en una cierta baja frecuencia en nuestra población ancestral, todavía podría ser correcta. Sin embargo, si esta hipótesis u otra que implica que la homosexualidad es adaptativa (o al menos no mal adaptativa) es falsa, no se sigue que la homosexualidad sea una enfermedad, ni siquiera si los homosexuales sufren en sociedades homofóbicas. La conjunción de causalidad por disfunción más deterioro no es suficiente para el trastorno, cuando el deterioro se debe a condiciones sociales que pueden alterarse con relativa facilidad; es decir, cuando las alteraciones necesarias para eliminar el deterioro no son alteraciones, tenemos buenas razones para negarnos a realizarlas (porque impondrían costos significativos a terceros, por ejemplo) [2]. Expreso esta afirmación diciendo que es una condición necesaria para que una condición sea una enfermedad que cause sufrimiento en casi cualquier entorno accesible. Si es el caso, hay un entorno accesible, donde la accesibilidad es una función no solo de la posibilidad física, sino también de los costos (económicos, sociales, morales) de acceder realmente a ese entorno, en los cuales una disfunción no causa un impedimento, entonces la disfunción no es suficiente para una enfermedad.
Al parecer, los contraejemplos aparentes de esta cuenta son aparentes. Considere la alergia al maní [3]. Ciertamente, es posible alterar el entorno de los enfermos de manera que no sufran ningún deterioro. Ese hecho implica que si tales alteraciones son lo suficientemente baratas, la alergia al maní no es una enfermedad. Esto parece ser contrario al uso médico estándar (ICD-10, por ejemplo, tiene una categoría adecuada para la alergia al maní). Sin embargo, a pesar del uso médico estándar, mantengo que si es cierto que existen entornos accesibles en los que una alergia al maní no causa ningún deterioro, entonces no es una enfermedad (quizás la intuición de que es una enfermedad se debe en parte al hecho de que evitar los cacahuetes en este momento esté lejos de ser gratuito, ya que la carga recae en los individuos de vigilar cuidadosamente su dieta en un entorno en el que muchos productos y platos contienen trazas de nueces suficientes para desencadenar la reacción alérgica. Comparar una alergia al maní a la dislexia. La dislexia puede tener una base genética, pero parece erróneo decir que nuestros ancestros cazadores-recolectores sufrían de dislexia antes de la invención de la escritura. Más bien, la dislexia parece ser una enfermedad solo en una sociedad en la que la lectura es lo suficientemente importante como para que los problemas de lectura cuenten como una discapacidad (Buchanan et al.,2000, p. 123). Ahora, si es cierto que la dislexia no fue una enfermedad en el pasado pre-alfabetizado, porque no causó un deterioro en aquellos que (en cierto sentido atenuado) la padecieron, entonces parece que si fuera posible modificarla sin costo alguno. El medio ambiente para que no causara un deterioro en los enfermos de hoy, no contaría como una enfermedad hoy. Sería análogo a la homosexualidad, en la medida en que nos correspondería eliminar el sufrimiento que causa al alterar el medio ambiente. El ejemplo de alergia al maní también parece ser muy similar, y por lo tanto sostengo que no constituye un contraejemplo de la cuenta ofrecida [4].
Según este relato, la adicción no es una enfermedad cerebral, porque a veces no es una enfermedad en absoluto. Si bien la afirmación de que el sistema dopaminérgico es disfuncional en los adictos es plausible, una disfunción de este tipo no es suficiente para el deterioro en muchos entornos accesibles. La tergiversación identificada es a nivel subpersonal, pero un agente sufre una patología de la mente solo cuando hay un problema a nivel personal. La enfermedad mental se identifica de manera bastante plausible con un defecto de racionalidad de algún tipo (Graham, 2010), y una tergiversación subpersonal no es un defecto de racionalidad. Es muy posible que los mecanismos se tergiversen mientras que los agentes representan adecuadamente; una vez que alguien se familiariza con una ilusión visual particular, esto puede ser cierto para ella en futuros encuentros con ella. Por supuesto, una tergiversación subpersonal podría en algunos casos causar directamente una tergiversación a nivel personal, pero eso no parece ser, directamente, el caso de la adicción. Si los agentes aceptaran la valoración de los medicamentos a los que son adictos a los mecanismos subpersonales, no querrían darse por vencidos, y una serie de datos sobre ellos serían inexplicables (por qué a menudo, aunque no siempre, dicen que quieren dar) arriba, por qué gastan recursos significativos en un aparente intento de rendirse (Ross et al., 2008); por qué la recuperación espontánea es tan común).
En la visión de Holton y Berridge (de próxima aparición), la adicción implica ansias intensas, así como tergiversaciones; Para ellos, el sistema de dopamina del cerebro medio es el sistema que genera los antojos en lugar de un sistema representativo. Esta adición a la cuenta podría ayudar de alguna manera a explicar cómo la adicción es un defecto a nivel personal: el agente experimenta estos antojos sin importar cómo juzga, y por lo tanto está motivado a actuar. Sin embargo, incluso con esta adición, parece que las disfunciones hipotéticas no llegan a ser una patología, porque existen entornos accesibles en los que el agente no sufrirá ningún defecto de racionalidad o deterioro de la agencia. Hay dos razones para esto. La primera es que las disfunciones identificadas no son suficientes para la experiencia de los antojos. Los antojos de drogas son muy dependientes de la señal,
En segundo lugar, aunque los antojos son desagradables, experimentarlos parece estar muy lejos de cualquier tipo de enfermedad mental o patología. La sobrevaluación subpersonal de las drogas más los antojos intensos no son suficientes para que la persona sufra un defecto de racionalidad. Tampoco son suficientes para que la persona sufra un deterioro suficientemente importante de la agencia o de su capacidad para llevar una vida que valga la pena. Las neuroadaptaciones características de la adicción son duraderas; Es por esta razón que el eslogan de Alcohólicos Anónimos «una vez que es alcohólico, siempre es alcohólico» tiene más que un grano de verdad. Sin embargo, claramente el ex bebedor o drogadicto que ha sido abstinente durante muchos años no tiene por qué sufrir ningún impedimento (aunque puede ser vulnerable a sufrir un impedimento). Todo por sí mismo, este hecho muestra que la disfunción neuropsicológica subyacente a la adicción no es suficiente para la enfermedad. De hecho, con respecto a algunas adicciones, algunos individuos que satisfacen la condición de disfunción no sufren deterioro a pesar de continuar tomando la droga. Si esto es cierto, variará según el medicamento, el método de consumo y (lo que es más importante) la capacidad del agente para acceder al medicamento de forma segura y confiable. La mayoría de las personas adictas a la cafeína no sufren deterioro. Más controvertido, algunos individuos adictos a las benzodiazepinas o a la nicotina liberada por el «cigarrillo electrónico» pueden no sufrir deterioro de la racionalidad, de la agencia o de la capacidad de perseguir una vida que valga la pena. Incluso algunos adictos a la heroína, con los recursos para obtener heroína de fuentes seguras,
No ayudará al defensor del concepto de enfermedad cerebral agregar los otros correlatos neuronales asociados con la adicción a la mezcla. Considere la desviación crónica del punto de ajuste de recompensa identificado por Koob y Le Moal (2008). El estado alostático que postulan es el resultado de la adaptación del cerebro a la ingesta de drogas. Mientras la droga esté disponible de manera confiable, la persona no sufrirá los efectos negativos de esta neuroadaptación [5]. Más bien, el estado anhedónico que sufren los individuos está asociado con la abstinencia crónica. Identificar la patología con este estado desagradable implica, contraintuitivamente, que el adicto abstinente sufre de una patología, pero el adicto que está usando no.
Otras neuroadaptaciones características de la adicción son los candidatos más plausibles para una patología con deterioro de la capacidad de la agencia. Se puede esperar que las disfunciones en los mecanismos involucrados en el autocontrol afecten la agencia en una variedad de condiciones. Sin embargo, en muchos entornos y para muchos individuos, el defecto no es tan significativo como para implicar un deterioro de la agencia o la racionalidad. Más bien, en entornos de apoyo, donde el agente está protegido de muchas demandas por el apoyo social, este impedimento es totalmente compatible con la búsqueda de una buena vida.
Si mi afirmación de que los correlatos neuronales de la adicción no causan deterioro en todos los entornos accesibles es cierta, la adicción no es una enfermedad cerebral en la forma en que el otro condiciona a Leshner (1997).) Las citas son. El accidente cerebrovascular, la esquizofrenia y la enfermedad de Alzheimer causan defectos significativos de racionalidad y dependencia en casi cualquier entorno; aunque podría ser posible imaginar entornos en los que algunas de estas condiciones no causaron deterioro, tales entornos no son realmente accesibles (para empezar, los costos de mantenerlos serían prohibitivos). La adicción difiere de las enfermedades cerebrales paradigmáticas en que sus correlatos no causan deterioro en todos los entornos, o en entornos casi accesibles. Para algunas condiciones que causan sufrimiento, los correlatos neurales son suficientes para causar un deterioro y para otros no lo son; solo aquellos que encajan en la clase anterior se consideran enfermedades cerebrales. La adicción encaja en la última clase porque,
Estas observaciones sugieren que si la adicción se entiende correctamente como una enfermedad o patología, no es solo porque se trata de una disfunción neuropsicológica. Más bien, capturar la manera en que es patológico requiere que adoptemos una explicación explícitamente normativa de la patología, según la cual alguien sufre de una patología cuando y solo cuando están sujetos a importantes impedimentos de agencia y, en consecuencia, de la capacidad de perseguir un bien. vida. En ausencia de tal deterioro, la persona que ingiere drogas y experimenta los cambios neuronales asociados con el uso de drogas a largo plazo no sufre de una enfermedad.
Los comentarios anteriores no deben ser una sorpresa: no son más que la afirmación de que la adicción debe encajar en el influyente modelo de enfermedad o trastorno en dos etapas. En el modelo de dos etapas, un individuo sufre de un trastorno solo si experimenta una disfunción biológica y esa disfunción es perjudicial, donde el juicio del daño se hace por referencia a las normas sociales de florecimiento (Wakefield, 1992; Murphy, 2006). La disfunción biológica puede ser una condición necesaria para ser un trastorno. Pero esta condición necesaria no es una condición suficiente, y la adicción no es una enfermedad cerebral. Más bien, cuando es una enfermedad, es una enfermedad que esencialmente involucra disfunción cerebral.
¿Por qué importa que la adicción no es una enfermedad cerebral?
La afirmación de que la enfermedad mental implica parcialmente, pero esencialmente, una cierta desviación de las normas no implica aceptar el modelo moral. No implica que los adictos sean los culpables de su adicción. Esto debería ser obvio, ya que la cuenta enfatiza que la adicción puede no contar como una enfermedad porque el sufrimiento que causa se debe en gran medida a las condiciones sociales que son, en cierto sentido, opcionales; Claramente la adicta no es ella misma responsable de estas condiciones. La explicación tampoco implica que la adicción no sea real, o que el sufrimiento involucrado no sea genuino. Puede haber un hecho del asunto si y cuando los adictos sufren un deterioro genuino de la agencia. Hay una variedad de relatos realistas de lo que constituye una buena vida. La adicción puede ser una falla normativa, pero si estas cuentas son correctas, no es una falla normativa. En lugar de un desorden natural explicable. Más bien, es una falla normativa debido a la clase de trastorno natural explicable que es.
Los comentarios anteriores son importantes, porque nos ayudan a reconocer que la insistencia de que la adicción es una enfermedad cerebral es solo una de las maneras en que podemos evitar tanto el burdo moralismo de quienes culpan a los adictos como un relativismo fácil sobre los trastornos. La adicción no es una enfermedad cerebral, pero hay un buen caso para decir que es, sin embargo, un trastorno que puede requerir tratamiento (que puede ser médico o psiquiátrico, aunque otros tipos de tratamiento pueden ser apropiados, además), para que el que sufre no tiene la culpa y el que sufre es un receptor apropiado de compasión.
Hasta ese punto, mi afirmación de que la adicción no es una enfermedad cerebral puede parecer que no cambia nada, en comparación con la situación que prevalecería si la afirmación de los científicos es que es una enfermedad cerebral que debe aceptarse. Aunque la superposición entre las dos cuentas es importante, existen algunas diferencias importantes.
La afirmación de que la adicción no es una enfermedad cerebral nos permite resituar al adicto en su entorno social (Levy, 2007). Ella sufre de un trastorno solo en la medida en que su cerebro es disfuncional de ciertas maneras y las condiciones sociales prevalecientes hacen que sea probable que ella sufra un defecto de racionalidad o un deterioro de la agencia como resultado. Esto puede deberse al hecho de que carece de los recursos para retirarse de los entornos en los que se encuentra con frecuencia con las señales que le provocan antojos, y en los que sus recursos de autocontrol se agotan por las constantes demandas, el estrés y la mala nutrición. Puede deberse al hecho de que carece de acceso a bienes que compiten con las atracciones de la droga. Los hechos que explican su adicción y los hechos que explican su sufrimiento (y el sufrimiento que causa a los demás) son en parte hechos sobre ella y en parte sobre el entorno en el que está incrustada. Además, los hechos sobre ella que explican su adicción y el sufrimiento asociado están mediados por su entorno (y algunos, y solo algunos) de los hechos sobre su entorno están mediados por ella).
Responder adecuadamente a la adicción, así como repartir culpas entre los adictos y otros actores, requiere que seamos sensibles a estos hechos [6]. La adicción es una patología que involucra disfunción neuropsicológica, y puede ser apropiado responder a ella tratando esta disfunción (farmacológicamente, por ejemplo). Pero la adicción es una patología solo debido al arraigo social de los adictos, y puede ser igualmente apropiado responder a ella alterando las condiciones sociales que la causan y la sostienen, o que causan y sostienen las deficiencias que genera. Si queremos entender la adicción y responder adecuadamente a ella, no debemos enfocarnos solo en la persona adicta, y mucho menos en su cerebro. Nuestro enfoque debe estar en ella, en su entorno social. Inevitablemente, eso implica que nosotros debemos ser sometidos a escrutinio; Tal vez necesitamos cambiar tanto como ella lo hace.
[1] Cabe señalar que el propio Leshner (1997, p. 46) reconoce que la adicción no es solo una enfermedad cerebral; más bien, afirma que es «una enfermedad cerebral para la cual los contextos sociales en los que ambos se han desarrollado y se expresan son de importancia crítica». Para Leshner, la adicción es una enfermedad cerebral en un contexto social, al igual que (para citar sus propios ejemplos) apoplejía, esquizofrenia y enfermedad de Alzheimer. Mi afirmación es que la adicción no es una enfermedad cerebral como las otras condiciones que cita Leshner; Tiene características cruciales que lo hacen diferente de apoplejía, esquizofrenia y Alzheimer.
[2] Sin embargo, supongamos que el deterioro es causado por una disfunción más condiciones sociales que no podemos alterar, o alterar, lo que sería prohibitivamente costoso, pero esas condiciones sociales son injustas. Supongamos, por ejemplo, que algún tipo de disfunción biológica causó un deterioro solo porque las personas fueron rechazadas por él; ¿tal condición contaría como una enfermedad (agradezco a Jerome Wakefield por plantear esta pregunta)? Creo que deberíamos considerar cualquier disfunción que cause un deterioro como una enfermedad cuando el deterioro no se puede evitar (por motivos prácticos). Esta vinculación de mi cuenta podría parecer contraria a la intuición; tal vez la impresión se pueda suavizar al señalar que en casos como este, podríamos deberle a los pacientes por el impedimento algún deber especial de recompensa o compensación.
[3] Gracias a Richard Holton por presionarme en este tema; La sugerencia de que la alergia al maní es un contraejemplo de la cuenta de la enfermedad que se ofrece aquí se debe a él.
[4] El ejemplo de la dislexia ilustra bien cómo la «accesibilidad» es una noción parcialmente normativa: un entorno cuenta como accesible si no es simplemente físicamente posible para un individuo que tiene acceso a él, o para que una sociedad entera adopte sus normas, pero es razonable Espere que la persona o sociedad tome estos pasos. Es posible que sigan existiendo culturas en las que la dislexia no cause un deterioro, porque la alfabetización no es un beneficio para los miembros. Sin embargo, para la gran mayoría de los enfermos, estas culturas son extremadamente difíciles de acceder: las barreras para ingresar a estas culturas (aprender un nuevo idioma y una nueva forma de vida) son altas, los recién llegados pueden no ser aceptados fácilmente y las personas originalmente están enculturadas en una cultura diferente. El modo de vida podría encontrar la nueva cultura muy insatisfactoria. Además, los miembros de estas culturas pueden ser alimentados inadecuadamente, puede carecer de acceso a agua potable limpia y de atención médica. Estos hechos implican que no sería razonable esperar que la mayoría de los disléxicos eviten el deterioro al acceder a estas culturas, incluso si existe algún sentido en el que algunos de ellos puedan hacerlo; por razones casi análogas, no sería razonable esperar que las sociedades desarrolladas adopten los modos de vida de tales culturas. Agradezco a Jerome Wakefield por presionarme en este tema.
[5] Debido a que la disponibilidad confiable de la droga permite al adicto mantener el equilibrio homeostático, la adicción parece ser un contraejemplo a la afirmación de Roe y Murphy (2011) de que es una condición necesaria para tener un trastorno que desordene los mecanismos diseñados para mantener la homeostasis. Un adicto puede sufrir un deterioro, causado por una disfunción del sistema dopaminérgico, y aun así ser capaz de mantener la homeostasis.
[6] La afirmación de que una respuesta adecuada a la adicción requiere el tratamiento del adicto en su contexto y, por lo tanto, abordar los factores sociales que no solo causan sufrimiento sino que también juegan un papel causal en el mantenimiento de la conducta, ha sido desarrollada con sensibilidad por Hanna Pickard ( ver Pickard, 2012 ; Pickard y Pearce, de próxima publicación)
Referencias
Baumeister R. F., Vohs K. D. (2007). Self-regulation, ego depletion, and motivation. Soc. Personal. Psychol. Compass 1, 1–1410.1111/j.1751-9004.2007.00008.x
Berridge K. C. (2007). The debate over dopamine’s role in reward: the case for incentive salience. Psychopharmacology (Berl.) 191, 391–43110.1007/s00213-006-0578-x
Booth M. (1996). Opium: A history. London: Simon and Schuster
Buchanan A., Brock D. W., Daniels N., Wikler D. (2000). From Chance to Choice: Genetics and Justice. Cambridge: Cambridge University Press
Carter A., Hall W. (2012). Addiction Neuroethics: The Promises and Perils of Neuroscience Research on Addiction. Cambridge: Cambridge University Press
Cosmides L., Tooby J. (1992). “Cognitive adaptations for social exchange,” in The Adapted Mind, eds Barkow J., Cosmides L., Tooby J., editors. (New York: Oxford University Press; ), 163–228
Cummins R. (1975). Functional analysis. J. Philos. 72, 741765.10.2307/2024640
Durrant R., Adamson S., Todd F., Sellman D. (2009). Drug use and addiction: an evolutionary perspective. Aust. N. Z. J. Psychiatry 43, 1049–105610.1080/00048670903270449
Murphy D. (2006). Psychiatry in the Scientific Image. Cambridge, MA: MIT Press [Google Scholar]
Murphy D., Stich S. (2000). “Darwin in the madhouse: evolutionary psychology and the classification of mental disorders,” in Evolution and the Human Mind: Modularity, Language and Meta-Cognition, eds Carruthers P., Chamberlain A., editors. (Cambridge: Cambridge University Press; ), 62–92
Nesse R. M., Williams G. C. (1995). Why We Get Sick. New York: Times Books
Roe K., Murphy D. (2011). “Function, dysfunction, and adaptation?” in Maladapting Minds: Philosophy, Psychiatry, and Evolutionary Theory, ed. Adriaens P. R., de Block A., editors. (Oxford: Oxford University Press; ), 216–237
Ross D., Sharp C., Vuchinich R. E., Spurrett D. (2008). Midbrain Mutiny: The Picoeconomics and Neuroeconomics of Disordered Gambling. Cambridge: MIT Press
Schultz W., Dayan P., Montague P. R. (1997). A neural substrate of prediction and reward. Science 275, 1593–115910.1126/science.275.5306.1593
Solinas M., Ferré S., You Z.-B., Karcz-Kubicha M., Popoli P., Goldberg S. R. (2002). Caffeine induces dopamine and glutamate release in the shell of the nucleus accumbens. J. Neurosci. 22, 6321–6324
Sullivan R. J., Hagen E. H. (2002). Psychotropic substance-seeking: evolutionary pathology or adaptation?Addiction 97, 389–34010.1046/j.1360-0443.2002.00024.x
Sutton R. S., Barto A. C. (1998). Reinforcement learning: An introduction. London: MIT Press
Volkow N. D., Li T. K. (2004). Drug addiction: the neurobiology of behaviour gone awry. Nat. Rev. Neurosci. 5, 963–97010.1038/nrn1539
Wakefield J. C. (1992). The concept of mental disorder: on the boundary between biological facts and social values. Am. Psychol. 47, 373–38810.1037/0003-066X.47.3.373
Wilson E. O. (1975). Sociobiology: The New Synthesis. Cambridge, MA: Harvard University Press
1Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, VIC, AustraliaEdited by: Hanna Pickard, University of Oxford, UKReviewed by: Richard Holton, Massachusetts Institute of Technology, USA; Jerome C. Wakefield, New York University, USA *Correspondence: Neil Levy, Florey Institute of Neuroscience and Mental Health, Royal Parade, The University of Melbourne, Parkville, VIC 3010, Australia. e-mail: ku.ca.xo.yhposolihp@yvel.lienThis article was submitted to Frontiers in Addictive Disorders and Behavioral Dyscontrol, a specialty of Frontiers in Psychiatry.
Alan I. Leshner fue director del Instituto Nacional sobre el Abuso de Drogas (NIDA) entre 1994 y 2001
Estados Unidos está atrapado en sus metáforas del abuso de drogas y en argumentos polarizados sobre ellas. Todos tienen una opinión. Un lado insiste en que debemos controlar la oferta, el otro en que debemos reducir la demanda. Las personas ven la adicción como una enfermedad o como un fracaso de la voluntad. Ninguno de estos análisis de calcomanías nos mueve hacia adelante. La verdad es que progresaremos en el tratamiento de los problemas de drogas solo cuando nuestro discurso nacional y nuestras estrategias sean tan complejas e integrales como el problema mismo.
Se logrará un mayor progreso contra el abuso de drogas cuando nuestras estrategias reflejen todas las complejidades de la última comprensión científica.
Un concepto central que ha ido evolucionando con los avances
científicos durante la última década es que la adicción a las drogas es una
enfermedad cerebral que se desarrolla con el tiempo como resultado del
comportamiento inicialmente voluntario del uso de drogas. La consecuencia es un
ansia compulsiva, búsqueda y uso de drogas prácticamente incontrolable que
interfiere con, si no destruye, el funcionamiento de un individuo en la familia
y en la sociedad. Esta condición médica exige tratamiento formal.
Ahora sabemos con gran detalle los mecanismos cerebrales a
través de los cuales las drogas modifican agudamente el estado de ánimo, la
memoria, la percepción y los estados emocionales. Usar drogas repetidamente a
lo largo del tiempo cambia la estructura y función del cerebro de manera
fundamental y duradera que puede persistir mucho después de que el individuo
deja de usarlas. La adicción se produce a través de una serie de cambios
neuroadaptativos y el establecimiento y fortalecimiento de nuevas conexiones de
memoria en varios circuitos en el cerebro. Todavía no conocemos todos los
mecanismos relevantes, pero la evidencia sugiere que esos cambios cerebrales
duraderos son responsables de las distorsiones del funcionamiento cognitivo y
emocional que caracterizan a los adictos, en particular la obligación de usar
drogas que es la esencia de la adicción. Es como si las drogas hubieran robado
los circuitos naturales de control motivacional del cerebro, dando como
resultado que el uso de drogas se convierta en la única, o al menos la
principal prioridad de motivación para el individuo. Por lo tanto, la mayoría
de la comunidad biomédica ahora considera que la adicción, en esencia, es una
enfermedad cerebral: una condición causada por cambios persistentes en la
estructura y función del cerebro.
Esta visión de la adicción basada en el cerebro ha generado una controversia sustancial, particularmente entre las personas que parecen capaces de pensar solo de manera polarizada. Muchas personas creen erróneamente que las explicaciones biológicas y de comportamiento son formas alternativas o competitivas de comprender los fenómenos, cuando en realidad son complementarias e integrables. La ciencia moderna ha enseñado que es demasiado simplista establecer la biología en oposición al comportamiento o enfrentar la fuerza de voluntad contra la química del cerebro. La adicción implica componentes biológicos y de comportamiento inseparables. Es el trastorno bioconductual por excelencia.
Muchas personas también creen erróneamente que la adicción a las drogas es simplemente un fracaso de voluntad o de fortaleza de carácter. La investigación contradice esa posición. Sin embargo, el reconocimiento de que la adicción es una enfermedad cerebral no significa que el adicto sea simplemente una víctima desafortunada. La adicción comienza con el comportamiento voluntario del uso de drogas, y los adictos deben participar y asumir una responsabilidad significativa por su recuperación. Por lo tanto, tener esta enfermedad cerebral no exime al adicto de la responsabilidad de su comportamiento, pero sí explica por qué un adicto no puede simplemente dejar de usar drogas solo por pura fuerza de voluntad. También dicta un enfoque mucho más sofisticado para lidiar con la variedad de problemas que rodean el abuso de drogas y la adicción en nuestra sociedad.
La esencia de la adicción
Todo el concepto de adicción ha sufrido mucho por la imprecisión y la concepción errónea. De hecho, si fuera posible, sería mejor comenzar de nuevo con un término nuevo y más neutral. La confusión se produce en parte debido a una distinción ahora arcaica entre si drogas específicas son adictivas «física» o «psicológicamente». La distinción históricamente giraba en torno a si los síntomas dramáticos de abstinencia física ocurren o no cuando un individuo deja de tomar un medicamento; lo que en el campo ahora llamamos «dependencia física».
Sin embargo, 20 años de investigación científica han enseñado que centrarse en esta distinción física versus psicológica está fuera de lugar y es una distracción de los problemas reales. Desde la perspectiva clínica y política, en realidad no importa mucho qué síntomas de abstinencia física ocurran. La dependencia física no es tan importante, porque incluso los síntomas dramáticos de abstinencia de la adicción a la heroína y al alcohol ahora se pueden manejar fácilmente con los medicamentos adecuados. Aún más importante, muchas de las drogas más peligrosas y adictivas, como la metanfetamina y la cocaína crack, no producen síntomas de dependencia física muy graves al momento de la abstinencia.
Lo que realmente importa más es si una droga causa o no lo que ahora sabemos que es la esencia de la adicción: falta de control, ansia compulsiva de drogas, búsqueda y uso, incluso frente a consecuencias negativas para la salud y sociales. Este es el quid de la forma en que el Instituto de Medicina, la Asociación Americana de Psiquiatría y la Asociación Médica Americana definen la adicción y cómo todos deberíamos usar el término. En realidad, solo esta calidad compulsiva de adicción es lo que importa a largo plazo para el adicto y para su familia, y eso debería ser importante para la sociedad en su conjunto. El deseo compulsivo que abruma a todas las demás motivaciones es la causa raíz de los problemas sociales y de salud masivos asociados con la adicción a las drogas. Al actualizar nuestro discurso nacional sobre el abuso de drogas, debemos tener en cuenta esta definición simple: La adicción es una enfermedad cerebral expresada en forma de comportamiento compulsivo. Tanto el desarrollo como la recuperación dependen de la biología, el comportamiento y el contexto social.
También es importante corregir la impresión errónea común de que el uso de drogas, el abuso y la adicción son puntos en un solo continuo a lo largo del cual uno se desliza hacia adelante y hacia atrás con el tiempo, pasando de usuario a adicto, luego de regreso a un usuario ocasional, luego de vuelta al adicto. La observación clínica y los estudios de investigación más formales respaldan la opinión de que, una vez adicto, el individuo ha pasado a un estado diferente de ser. Es como si se hubiera cruzado un umbral. Muy pocas personas parecen capaces de volver con éxito al uso ocasional después de haber sido realmente adictas. Desafortunadamente, todavía no tenemos un marcador biológico o de comportamiento claro de esa transición del uso voluntario de drogas a la adicción. Sin embargo, se está desarrollando rápidamente un conjunto de evidencia científica que apunta a una variedad de cambios celulares y moleculares en circuitos cerebrales específicos. Además, muchos de estos cambios cerebrales son comunes a todas las adicciones químicas, y algunos también son típicos de otros comportamientos compulsivos, como comer en exceso patológicamente.
La adicción debe entenderse como una enfermedad crónica recurrente. Aunque algunos adictos obtienen un control total sobre su consumo de drogas después de un solo episodio de tratamiento, muchos tienen recaídas. Los tratamientos repetidos se hacen necesarios para aumentar los intervalos y disminuir la intensidad de las recaídas, hasta que el individuo logre la abstinencia.
La complejidad de esta enfermedad cerebral no es atípica, porque prácticamente ninguna enfermedad cerebral es simplemente de naturaleza y expresión biológica. Todos, incluidos los accidentes cerebrovasculares, la enfermedad de Alzheimer, la esquizofrenia y la depresión clínica, incluyen algunos aspectos conductuales y sociales. Sin embargo, lo que puede hacer que la adicción parezca única entre las enfermedades cerebrales es que comienza con un comportamiento claramente voluntario: la decisión inicial de usar drogas. Además, no todos los que usan drogas se vuelven adictos. Los individuos difieren sustancialmente en la facilidad y rapidez en que se vuelven adictos y en sus preferencias por determinadas sustancias. De acuerdo con la naturaleza bioconductual de la adicción, estas diferencias individuales resultan de una combinación de factores ambientales y biológicos, particularmente genéticos De hecho, se estima que entre 50 y 70 por ciento de la variabilidad en la susceptibilidad a volverse adicto puede explicarse por factores genéticos.
Aunque las características genéticas pueden predisponer a los individuos a ser más o menos susceptibles de volverse adictos, los genes no condenan a uno a convertirse en adictos.
Con el tiempo, el adicto pierde un control sustancial sobre su comportamiento inicialmente voluntario y se vuelve compulsivo. Para muchas personas, estos comportamientos son verdaderamente incontrolables, al igual que la expresión conductual de cualquier otra enfermedad cerebral. Los esquizofrénicos no pueden controlar sus alucinaciones y delirios. Los pacientes de Parkinson no pueden controlar su temblor. Los pacientes clínicamente deprimidos no pueden controlar voluntariamente su estado de ánimo. Por lo tanto, una vez que uno es adicto, las características de la enfermedad, y los enfoques de tratamiento, no son tan diferentes de la mayoría de las otras enfermedades cerebrales. No importa cómo se desarrolle una enfermedad, una vez que la tiene, uno está enfermo y necesita tratamiento.
Además, los patrones de comportamiento voluntarios están, por supuesto, involucrados en la etiología y la progresión de muchas otras enfermedades, aunque no todas las enfermedades cerebrales. Abundan los ejemplos, que incluyen hipertensión, arteriosclerosis y otras enfermedades cardiovasculares, diabetes y formas de cáncer en las que el inicio está fuertemente influenciado por la alimentación, el ejercicio, el tabaquismo y otros comportamientos del individuo.
Los comportamientos adictivos tienen características especiales relacionadas con los contextos sociales en los que se originan. Todos los estímulos ambientales que rodean el uso inicial de drogas y el desarrollo de la adicción en realidad se «condicionan» a ese uso de drogas y, por lo tanto, son críticos para el desarrollo y la expresión de la adicción. Los estímulos ambientales se combinan a tiempo con las experiencias iniciales de uso de drogas de un individuo y, a través del condicionamiento clásico, adquieren propiedades de estímulo condicionados. Cuando esos estímulos están presentes en un momento posterior, provocan la anticipación de una experiencia de drogas y, por lo tanto, generan un ansia tremenda de drogas. El ansia inducida por estímulos es una de las causas más frecuentes de recaídas en el consumo de drogas, incluso después de largos períodos de abstinencia, independientemente de si hay drogas disponibles.
La importancia de los estímulos ambientales o contextuales ayuda a explicar por qué el reingreso a la comunidad puede ser tan difícil para los adictos que abandonan los entornos controlados de tratamiento o entornos correccionales y por qué el cuidado posterior es tan esencial para una recuperación exitosa. La persona que se convirtió en adicta en el hogar está constantemente expuesta a los estímulos condicionados a su consumo inicial de drogas, como el vecindario donde pasó el rato, los amigos que consumen drogas o el lugar donde compró drogas. La simple exposición a esos estímulos desencadena automáticamente el deseo y puede conducir rápidamente a recaídas. Esta es una razón por la cual alguien que aparentemente superó los antojos de drogas mientras estaba en prisión o en un tratamiento residencial podría volver rápidamente al uso de drogas al regresar a casa. De hecho, uno de los objetivos principales del tratamiento de la drogadicción es enseñar a los adictos cómo lidiar con los tirones causados por la exposición inevitable a estos estímulos condicionados.
Trascendencia
Entender la adicción como una enfermedad cerebral tiene implicaciones amplias y significativas para la percepción pública de los adictos y sus familias, para la práctica del tratamiento de la adicción y para algunos aspectos de las políticas públicas. Por otro lado, esta visión biomédica de la adicción no habla directamente y es improbable que afecte significativamente a muchos otros temas, incluidas las estrategias específicas para controlar el suministro de drogas y si el uso inicial de drogas debe ser legal o no. Además, el modelo de adicción a la enfermedad cerebral no aborda la cuestión de si las drogas específicas de abuso también pueden ser medicamentos potenciales. Abundan los ejemplos de medicamentos que pueden ser medicamentos altamente adictivos y extremadamente efectivos. El ejemplo más conocido es el uso apropiado de la morfina como tratamiento para el dolor. Sin embargo, se pueden extraer varias lecciones prácticas de la comprensión científica de la adicción.
No es de extrañar que los adictos no puedan dejar de consumir solos. Tienen una enfermedad que requiere tratamiento biomédico. Las personas a menudo asumen que debido a que la adicción comienza con un comportamiento voluntario y se expresa en forma de comportamiento excesivo, las personas deberían poder dejar de consumir por la fuerza de la voluntad. Sin embargo, es esencial entender cuando se trata de adictos que estamos tratando con personas cuyos cerebros han sido alterados por el consumo de drogas. Necesitan tratamiento de adicción a las drogas. Sabemos que, contrariamente a la creencia común, muy pocos adictos se detienen por sí solos. Al observar que hay muy pocos adictos a la heroína en sus 50 o 60 años, las personas con frecuencia preguntan qué les sucedió a los que eran adictos a la heroína hace 30 años, suponiendo que debían haber dejado de consumir por su cuenta. Sin embargo, los estudios longitudinales encuentran que solo una fracción muy pequeña se rinde por sí sola. El resto ha sido tratado con éxito, actualmente se encuentra en tratamiento de mantenimiento o (aproximadamente la mitad) está muerto. Considere el ejemplo de fumar cigarrillos: varios estudios han encontrado que entre el 3 y el 7 por ciento de las personas que intentan dejar de fumar por sí mismas cada año en realidad tienen éxito. La ciencia finalmente ha convencido al público de que la depresión no es solo mucha tristeza; que las personas deprimidas se encuentran en un estado cerebral diferente y, por lo tanto, requieren tratamiento para controlar sus síntomas. Lo mismo es cierto para los pacientes esquizofrénicos. Es hora de reconocer que este también es el caso de los adictos.
El papel de la responsabilidad personal no se reduce, pero se aclara. ¿Tener una enfermedad cerebral significa que las personas adictas ya no tienen ninguna responsabilidad por su comportamiento o que simplemente son víctimas de su propia genética y química cerebral? Por supuesto no. La adicción comienza con el comportamiento voluntario del uso de drogas, y aunque las características genéticas pueden predisponer a los individuos a ser más o menos susceptibles a volverse adictos, los genes no condenan a uno a convertirse en adictos. Esta es una de las principales razones por las cuales los esfuerzos para prevenir el uso de drogas son tan vitales para cualquier estrategia integral para enfrentar los problemas de drogas de la nación. El consumo inicial de drogas es un comportamiento voluntario y, por lo tanto, prevenible.
Además, como con cualquier enfermedad, el comportamiento se convierte en una parte crítica de la recuperación. Como mínimo, uno debe cumplir con el régimen de tratamiento, que es más difícil de lo que parece. El cumplimiento del tratamiento es la principal causa de recaídas en todas las enfermedades crónicas, como el asma, la diabetes, la hipertensión y la adicción. Además, las tasas de cumplimiento del tratamiento no son peores para la adicción que para estas otras enfermedades, que van del 30 al 50 por ciento. Por lo tanto, tanto para la drogadicción como para otras enfermedades crónicas, la motivación y el comportamiento del individuo son claramente partes importantes del éxito en el tratamiento y la recuperación.
Implicaciones para los enfoques de tratamiento y las expectativas de tratamiento. Mantener esta comprensión bioconductual integral de la adicción también habla de lo que debe proporcionarse en los programas de tratamiento de drogas. Nuevamente, debemos tener cuidado de no enfrentar a la biología con el comportamiento. Los Principios para el Tratamiento Eficaz de la Drogadicción, recientemente publicados por el Instituto Nacional sobre el Abuso de Drogas (NIDA), brindan una discusión detallada sobre cómo debemos tratar todos los aspectos del individuo, no solo el componente biológico o el componente conductual. Al igual que con otras enfermedades cerebrales como la esquizofrenia y la depresión, los datos muestran que los mejores enfoques de tratamiento de adicción a las drogas atienden a todo el individuo, combinando el uso de medicamentos, terapias conductuales y atención a los servicios sociales y rehabilitación necesarios. Estos pueden incluir servicios tales como terapia familiar para permitir que el paciente regrese a una vida familiar exitosa, servicios de salud mental, educación y capacitación vocacional y servicios de vivienda. Eso no significa, por supuesto, que todas las personas necesiten todos los componentes del tratamiento y todos los servicios de rehabilitación. Otro principio del tratamiento eficaz de la adicción es que la variedad de servicios incluidos en el plan de tratamiento de un individuo debe coincidir con su conjunto particular de necesidades. Además, dado que esas necesidades seguramente cambiarán en el transcurso de la recuperación, la gama de servicios prestados deberá ser reevaluada y ajustada continuamente.
La entrada en el tratamiento de drogas no necesita ser completamente voluntaria para que funcione
Qué hacer con los delincuentes adictos. Una conclusión obvia es que debemos dejar de ver de manera simplista los enfoques de justicia penal y salud como opuestos incompatibles. La realidad práctica es que la delincuencia y la adicción a las drogas a menudo ocurren en conjunto: entre el 50 y el 70 por ciento de los detenidos son adictos a las drogas ilegales. Pocos ciudadanos estarían dispuestos a renunciar al control del sistema de justicia penal sobre las personas, ya sean adictas o no, que hayan cometido crímenes contra otros. Además, una amplia experiencia de la vida real muestra que si simplemente encarcelamos a los delincuentes adictos sin tratarlos, su retorno al uso de drogas y la criminalidad está prácticamente garantizado.
Un creciente cuerpo de evidencia científica apunta a un enfoque combinado de salud pública / seguridad pública mucho más racional y efectivo para tratar con el delincuente adicto. En resumen, los datos muestran que si los delincuentes adictos reciben un tratamiento bien estructurado de drogas mientras están bajo el control de la justicia penal, sus tasas de reincidencia pueden reducirse entre un 50 y un 60 por ciento para el uso posterior de drogas y en más del 40 por ciento para un comportamiento criminal adicional. Además, el ingreso al tratamiento farmacológico no necesita ser completamente voluntario para que funcione. De hecho, los estudios sugieren que una mayor presión para permanecer en el tratamiento, ya sea del sistema legal o de los miembros de la familia o los empleadores, en realidad aumenta la cantidad de tiempo que los pacientes permanecen en el tratamiento y mejora sus resultados.
Resultados como estos son la base de una tendencia muy importante en las estrategias de control de drogas que ahora se están implementando en los Estados Unidos y en muchos países extranjeros. Por ejemplo, alrededor del 40 por ciento de las prisiones en este país ahora afirman proporcionar algún tipo de tratamiento de drogas a sus reclusos adictos, aunque no sabemos la calidad del tratamiento brindado. El desvío a los programas de tratamiento de drogas como alternativa al encarcelamiento está ganando popularidad en todo Estados Unidos. El crecimiento ampliamente aplaudido en los tribunales de tratamiento de drogas en los últimos cinco años, a más de 400, es otro ejemplo exitoso de la combinación de enfoques de salud pública y seguridad pública. Estos tribunales de drogas usan una combinación de sanciones de justicia penal y herramientas de monitoreo y tratamiento del uso de drogas para manejar a los delincuentes adictos.
Actualizando la discusión
Comprender el abuso y la adicción a las drogas en toda su complejidad exige que superemos el pensamiento polarizado simplista sobre los problemas de las drogas. La adicción es tanto un problema de salud pública como de seguridad pública, no uno u otro. Debemos tratar los problemas de oferta y demanda con igual vigor. El abuso de drogas y la adicción tienen que ver con la biología y el comportamiento. Uno puede tener una enfermedad y no ser una víctima desafortunada de ella.
También debemos abandonar nuestra atracción por las metáforas simplistas que solo nos distraen del desarrollo de estrategias apropiadas. Yo, por mi parte, lamentaré de alguna manera ver desaparecer la metáfora de la Guerra contra las Drogas, pero desaparecer, debe hacerlo. En algún nivel, la noción de librar una guerra es tan apropiada para la enfermedad de la adicción como lo es para nuestra Guerra contra el Cáncer, lo que simplemente significa poner todas las fuerzas para enfrentar el problema de una manera enfocada y energizada. Pero, lamentablemente, este concepto se ha distorsionado y mal utilizado con el tiempo, y la Guerra contra las Drogas nunca se convirtió en lo que debería haber sido: la Guerra contra el Abuso y la Adicción a las Drogas. Además, preocuparse por si estamos ganando o perdiendo esta guerra se ha deteriorado al usar medidas simplistas e inapropiadas, como contar a los drogadictos. Al final, solo ha alimentado la discordia.
Sin embargo, espero que todos resistamos la tentación de reemplazarlo con otra frase pegadiza que inevitablemente se convertirá en una búsqueda de soluciones rápidas o fáciles de resolver para nuestros problemas de drogas. No confiamos en metáforas o estrategias simples para tratar nuestros otros problemas nacionales importantes, como la educación, la atención médica o la seguridad nacional. Después de todo, estamos tratando de resolver problemas verdaderamente monumentales y multidimensionales a escala nacional o incluso internacional. Devaluarlos al nivel de los eslóganes hace a nuestro público una injusticia y nos condena al fracaso.
Comprender los aspectos de salud de la adicción no es de ninguna manera incompatible con la necesidad de controlar el suministro de drogas. De hecho, un enfoque de salud pública para detener una epidemia o propagación de una enfermedad siempre se enfoca de manera integral en el agente, el vector y el huésped. En el caso de las drogas de abuso, el agente es la droga, el anfitrión es el abusador o el adicto, y el vector para transmitir la enfermedad es claramente los proveedores y vendedores de drogas que mantienen al agente fluyendo tan rápidamente. La prevención y el tratamiento son las estrategias para ayudar a proteger al huésped. Pero, así como debemos lidiar con las moscas y los mosquitos que transmiten enfermedades infecciosas, debemos abordar directamente todos los vectores en el sistema de suministro de drogas.
Para ser verdaderamente efectivos, los enfoques combinados de salud pública / seguridad pública propugnados aquí deben implementarse en todos los niveles de la sociedad: local, estatal y nacional. Todos los problemas de drogas son, en última instancia, de carácter e impacto local, ya que difieren mucho entre entornos geográficos y contextos culturales, y las soluciones más efectivas se implementan a nivel local. Cada comunidad debe trabajar a través de sus propias estrategias de implementación de antidrogas localmente apropiadas, y esas estrategias deben ser tan integrales y basadas en la ciencia como las instituidas a nivel estatal o nacional.
El mensaje del ahora muy amplio y profundo conjunto de evidencia científica es absolutamente claro. Si nosotros, como sociedad, esperamos hacer algún progreso real en el tratamiento de nuestros problemas de drogas, tendremos que superar la indignación moral de que los adictos se lo han «hecho a sí mismos» y desarrollar estrategias que sean tan sofisticadas y complejas como el problema en sí mismo. Si los adictos son «víctimas» o no, una vez adictos deben ser vistos como «pacientes con enfermedades cerebrales».
Además, aunque nuestras tradiciones nacionales abogan por la compasión por aquellos que están enfermos, no importa cómo contrajeron sus enfermedades, reconozco que muchos adictos han alterado no solo sus propias vidas sino también las de sus familias y sus comunidades más amplias, y por lo tanto no generan fácilmente compasión. Sin embargo, no importa cómo uno se sienta acerca de los adictos y sus historias de comportamiento, una gran cantidad de evidencia científica muestra que abordar la adicción como una enfermedad tratable es extremadamente rentable, tanto financieramente como en términos de impactos sociales más amplios, como la violencia familiar, el crimen y otras formas de agitación social. Por lo tanto, está claramente en el interés de todos superar el dolor y la indignación y frenar el consumo de drogas en la sociedad al mejorar los esfuerzos de prevención del uso de drogas y proporcionar tratamiento a todos los que lo necesitan.
Lectura recomendada
J. D. Berke and S. E. Hyman, “Addiction, Dopamine, and the Molecular Mechanisms of Memory,” Neuron 25 (2000): 515532 ).
H. Garavan, J. Pankiewicz, A. Bloom, J. K. Cho, L. Sperry, T. J. Ross, B. J. Salmeron, R. Risinger, D. Kelley, and E. A. Stein, “Cue-Induced Cocaine Craving: Neuroanatomical Specificity for Drug Users and Drug Stimuli,” American Journal of Psychiatry 157 (2000): 17891798 ).
I. Leshner, “Science-Based Views of Drug Addiction and Its Treatment,” Journal of the American Medical Association 282 (1999): 13141316 ).
T. McLellan, D. C. Lewis, C. P. O’Brien, and H. D. Kleber, “Drug Dependence, a Chronic Medical Illness,” Journal of the American Medical Association 284 (2000): 16891695 ).
National Institute on Drug Abuse, Principles of Drug Addiction Treatment: A Research-Based Guide (National Institutes of Health, Bethesda, MD, July 2000) ).
National Institute on Drug Abuse, Preventing Drug Use Among Children and Adolescents: A Research-Based Guide(National Institutes of Health, Bethesda, MD, March 1997) ).
Physician Leadership on National Drug Policy, position paper on drug policy (PLNDP Program Office, Brown University, Center for Alcohol and Addiction Studies, Providence, R.I.: January 2000) ).
F. S. Taxman and J. A. Bouffard, “The Importance of Systems in Improving Offender Outcomes: New Frontiers in Treatment Integrity,” Justice Research and Policy 2 (2000): 3758.
Los avances científicos en los últimos 20 años han demostrado que la adicción a las drogas es una enfermedad crónica y recurrente que resulta de los efectos prolongados de las drogas en el cerebro. Al igual que con muchas otras enfermedades cerebrales, la adicción tiene aspectos del contexto social y del comportamiento que son partes importantes del trastorno en sí. Por lo tanto, los enfoques de tratamiento más efectivos incluirán componentes biológicos, de comportamiento y de contexto social. Reconocer la adicción como un trastorno cerebral crónico y recurrente caracterizado por la búsqueda y el uso compulsivo de drogas puede afectar las estrategias de salud y políticas sociales generales de la sociedad y ayudar a disminuir los costos sociales y de salud asociados con el abuso de drogas y la adicción.
Los espectaculares avances en las últimas dos décadas, tanto
en las neurociencias como en las ciencias del comportamiento, han revolucionado
nuestra comprensión del abuso de drogas y la adicción. Los científicos han
identificado circuitos neuronales que subsumen las acciones de todas las drogas
conocidas de abuso y han especificado vías comunes que se ven afectadas por
casi todas esas drogas. Los investigadores también han identificado y clonado
los principales receptores para prácticamente todas las drogas de que se pueden
abusar, así como los enlaces naturales para la mayoría de esos receptores.
Además, han elaborado muchos de los procesos bioquímicos dentro de la célula
que persiguen la activación del receptor por parte de estas sustancias. La
investigación también ha comenzado a revelar diferencias importantes entre los
cerebros de individuos adictos y no adictos e indicar algunos elementos comunes
de la adicción, independientemente de la sustancia.
Esa es la buena noticia. La mala noticia es el dramático retraso entre estos avances en la ciencia y su apreciación por parte del público en general o su aplicación en la práctica o en la configuración de políticas públicas. Existe una gran brecha entre los hechos científicos y las percepciones públicas sobre el abuso de drogas y la adicción. Por ejemplo, muchas personas, quizás la mayoría, ven el abuso de drogas y la adicción como problemas sociales, que deben manejarse solo con soluciones sociales, particularmente a través del sistema de justicia penal. Por otro lado, la ciencia ha enseñado que el abuso de drogas y la adicción son tanto problemas de salud como problemas sociales. La consecuencia de esta brecha es un retraso significativo en ganar control sobre el problema del abuso de drogas.
Parte del retraso y la desconexión resultante provienen de la demora normal en la transferencia de cualquier conocimiento científico a la práctica y la política. Sin embargo, hay otros factores exclusivos del campo del abuso de drogas que complican el problema. Una barrera importante es el tremendo estigma asociado al uso de drogas o, peor aún, a un adicto. La opinión pública más benéfica de los drogadictos es como víctimas de su situación social. Sin embargo, la opinión más común es que los drogadictos son personas débiles o malas, que no están dispuestas a llevar una vida moral y controlar su comportamiento y gratificaciones. Por el contrario, la adicción es en realidad una enfermedad crónica y recurrente, caracterizada por la búsqueda y el uso compulsivo de drogas (1). El abismo en las implicaciones entre la visión de «persona mala» y la visión de «paciente con enfermedad crónica» es tremendo. Como un solo ejemplo, hay muchas personas que creen que las personas adictas ni siquiera merecen tratamiento. Este estigma, y el tono moralista subyacente, es una superposición significativa en todas las decisiones que se relacionan con el uso y los consumidores de drogas.
Otra barrera es que algunas de las personas que trabajan en el campo de la prevención del abuso de drogas y el tratamiento de la adicción también tienen ideologías arraigadas que, aunque suelen ser diferentes en su origen y forma a las ideologías del público en general, pueden ser igualmente problemáticas. Por ejemplo, muchos trabajadores del abuso de drogas son consumidores de drogas que tuvieron experiencias exitosas de tratamiento con un método de tratamiento particular. Por lo tanto, pueden defender celosamente un enfoque único, incluso ante la evidencia científica contradictoria. De hecho, hay muchos tratamientos para el abuso de drogas que han demostrado ser eficaces a través de ensayos clínicos (1, 2).
A pesar de estas dificultades, creo que podemos y debemos salvar esta desconexión informativa si vamos a hacer un progreso real en el control del abuso de drogas y la adicción. Es hora de reemplazar la ideología con la ciencia.
El abuso de drogas y la adicción como problemas de salud pública
En el nivel más general, las investigaciones han demostrado que el abuso de drogas es un problema de salud de doble filo, así como un problema social. Afecta tanto la salud del individuo como la salud pública. El uso de drogas tiene consecuencias negativas bien conocidas y graves para la salud, tanto mentales como físicas. Pero el abuso y la adicción a las drogas también tienen enormes implicaciones para la salud pública, porque el consumo de drogas, directa o indirectamente, es ahora un vector importante para la transmisión de muchas enfermedades infecciosas graves, en particular el síndrome de inmunodeficiencia adquirida (SIDA), la hepatitis y la tuberculosis. —Así como la violencia. Debido a que la adicción es un problema de salud tan complejo y generalizado, debemos incluir en nuestras estrategias generales un enfoque de salud pública comprometido, que incluya educación extensa y esfuerzos de prevención, tratamiento e investigación.
La ciencia está proporcionando la base para tales enfoques de salud pública. Por ejemplo, dos grandes conjuntos de estudios (3) han demostrado la efectividad de estrategias bien delineadas de alcance para modificar los comportamientos de individuos adictos que los ponen en riesgo de adquirir el virus de la inmunodeficiencia humana (VIH), incluso si continúan consumiendo drogas y no quieren entrar en tratamiento. Este enfoque es contrario a la opinión generalizada de que los adictos están tan incapacitados por las drogas que no pueden modificar ninguno de sus comportamientos. También sugiere una base para estrategias mejoradas para reducir las consecuencias negativas para la salud del uso de drogas inyectables para el individuo y para la sociedad.
Lo que importa en la adicción
La investigación científica y la experiencia clínica nos han enseñado mucho sobre lo que realmente importa en la adicción y dónde necesitamos concentrar nuestros esfuerzos clínicos y políticos. Sin embargo, con demasiada frecuencia, la atención se centra en los aspectos equivocados de la adicción, y los esfuerzos para abordar este problema difícil pueden estar mal orientados.
Cualquier discusión sobre las drogas psicoactivas se refiere inevitablemente a la cuestión de si una droga en particular es física o psicológicamente adictiva. En esencia, este problema gira en torno a si los síntomas de abstinencia física dramáticos ocurren o no cuando una persona deja de tomar una sustancia psicoactiva, lo que los profesionales en el campo suelen denominar dependencia física. El supuesto que a menudo se sigue es que cuanto más dramáticos sean los síntomas físicos de abstinencia, más grave o peligrosa debe ser la droga.
Este pensamiento está desactualizado. Tanto desde el punto de vista clínico como desde el punto de vista político, no importa mucho qué síntomas físicos de abstinencia se produzcan, si es que ocurren. En primer lugar, incluso los síntomas de abstinencia de la adicción a la heroína ahora se pueden controlar fácilmente con los medicamentos adecuados. Segundo, y más importante, muchas de las drogas más adictivas y peligrosas no producen síntomas físicos graves al abstenerse. La cocaína crack y la metanfetamina son ejemplos claros: ambos son altamente adictivos, pero el cese de su uso produce pocos síntomas físicos de abstinencia, ciertamente nada como los síntomas físicos que acompañan a la abstinencia de alcohol o heroína.
Lo que importa tremendamente es si una droga causa o no lo que ahora sabemos que es la esencia de la adicción: la búsqueda y el consumo compulsivo de sustancias psicoactivas, incluso ante consecuencias sociales y de salud negativas (4). Estas son las características que, en última instancia, son más importantes para el paciente y son donde deben dirigirse los esfuerzos de tratamiento. Estos comportamientos también son los elementos responsables de los problemas masivos de salud y sociales que trae consigo la adicción a las drogas.
La adicción es una enfermedad cerebral
Si bien cada sustancia que se ha estudiado tiene algunos mecanismos de acción idiosincrásicos, prácticamente todas las drogas de abuso tienen efectos comunes, ya sea directa o indirectamente, en una única vía profunda dentro del cerebro. Esta vía, el sistema de recompensa mesolímbico, se extiende desde el área tegmental central [1] hasta el núcleo accumbens [2], con proyecciones a áreas como el sistema límbico y la corteza orbitofrontal. La activación de este sistema parece ser un elemento común en lo que mantiene a los consumidores que toman drogas. Esta actividad no es exclusiva de ninguna droga; todas las sustancias adictivas afectan a este circuito (5).
El uso agudo de drogas no solo modifica la función cerebral de manera crítica, sino que el uso prolongado de drogas causa cambios generalizados en la función cerebral que persisten mucho después de que el individuo deja de tomar la droga. Se han identificado efectos significativos del uso crónico para muchas drogas en todos los niveles: molecular, celular, estructural y funcional (6, 7). El cerebro adicto es claramente diferente del cerebro no adicto, como lo manifiestan los cambios en la actividad metabólica del cerebro, la disponibilidad de receptores, la expresión de genes y la capacidad de respuesta a las señales ambientales. Algunos de estos cambios cerebrales de larga duración son idiosincrásicos a sustancia específicas, mientras que otros son comunes a muchas drogas diferentes (6–9). Los efectos cerebrales comunes de las sustancias adictivas sugieren mecanismos cerebrales comunes subyacentes a todas las adicciones (5, 7, 9, 10).
Esa adicción está vinculada a los cambios en la estructura y función del cerebro, lo que la convierte, fundamentalmente, en una enfermedad cerebral. Un interruptor metafórico en el cerebro parece ser arrojado como resultado del uso prolongado de drogas. Inicialmente, el uso de drogas es un comportamiento voluntario, pero cuando se produce ese cambio, el individuo pasa al estado de adicción, caracterizado por la búsqueda y el uso compulsivo de drogas (11).
Comprender que la adicción es, en esencia, una consecuencia de cambios fundamentales en la función cerebral significa que un objetivo importante del tratamiento debe ser revertir o compensar esos cambios cerebrales. Estos objetivos se pueden lograr a través de medicamentos o tratamientos conductuales [los tratamientos conductuales han tenido éxito en alterar la función cerebral en otros trastornos psicobiológicos (12)]. La aclaración de la biología subyacente al cambio metafórico es clave para el desarrollo de tratamientos más efectivos, en particular los medicamentos antiadicción.
Pero no solo una enfermedad cerebral
Por supuesto, la adicción no es tan simple. La adicción no es solo una enfermedad cerebral. Es una enfermedad cerebral para la cual los contextos sociales en los que se ha desarrollado y se expresa son de importancia crítica. El caso de los muchos miles de veteranos de la guerra de Vietnam que regresaron y que fueron adictos a la heroína ilustra este punto. En contraste con los adictos en las calles de los Estados Unidos, fue relativamente fácil tratar las adicciones de los veteranos que regresan. Este éxito fue posible porque se habían vuelto adictos mientras se encontraban en un entorno casi totalmente diferente al que habían regresado. En su hogar en los Estados Unidos, estuvieron expuestos a algunas de las señales ambientales condicionadas que inicialmente se habían asociado con su uso de drogas en Vietnam. La exposición a señales condicionadas puede ser un factor importante para causar antojos de drogas persistentes o recurrentes y recaídas en el uso de drogas incluso después de un tratamiento exitoso (13).
Las implicaciones son obvias. Si entendemos la adicción como una enfermedad psicobiológica prototípica, con componentes críticos de contexto biológico, conductual y social, nuestras estrategias de tratamiento deben incluir elementos biológicos, conductuales y de contexto social. No solo se debe tratar la enfermedad cerebral subyacente, sino que también deben abordarse los componentes de la señal conductual y social, al igual que ocurre con muchas otras enfermedades cerebrales, como los accidentes cerebrovasculares, la esquizofrenia y la enfermedad de Alzheimer.
Un trastorno crónico y recidivante
La adicción rara vez es una enfermedad aguda. Para la mayoría de las personas, es un trastorno crónico y recurrente. La abstinencia total por el resto de la vida es un resultado relativamente raro de un solo episodio de tratamiento. Las recaídas son más la norma. Por lo tanto, la adicción debe abordarse más como otras enfermedades crónicas, como la diabetes y la hipertensión crónica, que como una enfermedad aguda, como una infección bacteriana o un hueso roto (1). Este requisito tiene tremendas implicaciones sobre cómo evaluamos la efectividad del tratamiento y los resultados del tratamiento. Ver la adicción como un trastorno crónico y recurrente significa que un buen resultado del tratamiento, y la expectativa más razonable, es una disminución significativa en el uso de drogas y largos períodos de abstinencia, con solo recaídas ocasionales. Eso constituye un estándar razonable para el éxito del tratamiento, como es el caso de otras enfermedades crónicas, el manejo de la enfermedad, no una cura (1, 2).
Conclusión
La adicción como una enfermedad crónica y recidivante del cerebro es un concepto totalmente nuevo para gran parte del público en general, para muchos políticos y, lamentablemente, para muchos profesionales de la salud. Muchas de las implicaciones se han discutido anteriormente, pero hay otras.
A nivel de políticas, comprender la importancia del uso de drogas y la adicción tanto para la salud de las personas como para la salud del público afecta muchas de nuestras estrategias generales de salud pública. Una comprensión precisa de la naturaleza del abuso de drogas y la adicción también debe afectar nuestras estrategias de justicia penal. Por ejemplo, si sabemos que los delincuentes son drogadictos, ya no es razonable simplemente encarcelarlos. Si tienen una enfermedad cerebral, encarcelarlos sin tratamiento es inútil. Si no se los trata, sus tasas de reincidencia tanto para el crimen como para el uso de drogas son terriblemente altas; sin embargo, si los delincuentes adictos reciben tratamiento mientras están en prisión, ambos tipos de reincidencia pueden reducirse drásticamente (14). Por lo tanto, es contraproducente no tratar a los adictos mientras están en prisión.
A un nivel aún más general, entender la adicción como una enfermedad cerebral también afecta la forma en que la sociedad se acerca y trata con los individuos adictos. Tenemos que enfrentar el hecho de que incluso si la condición se produce inicialmente debido a un comportamiento voluntario (uso de drogas), el cerebro de un adicto es diferente del cerebro de un no adicto, y el individuo adicto debe ser tratado como si estuviera en un Estado del cerebro diferente. Hemos aprendido a tratar con personas en diferentes estados cerebrales para la esquizofrenia y la enfermedad de Alzheimer. Recuerde que recién a principios de este siglo todavía estábamos colocando a individuos con esquizofrenia en asilos como prisioneros, mientras que ahora sabemos que requieren tratamientos médicos. Ahora necesitamos ver al adicto como alguien cuya mente (leer: cerebro) ha sido alterada fundamentalmente por las drogas. El tratamiento es necesario para tratar la función cerebral alterada y los componentes concomitantes del comportamiento y el funcionamiento social de la enfermedad.
Entender la adicción como una enfermedad cerebral explica en parte por qué las estrategias políticas históricas que se centran únicamente en los aspectos de justicia penal o social del uso de drogas y la adicción no han tenido éxito. Les falta al menos la mitad del problema. Si el cerebro es el núcleo del problema, atender al cerebro debe ser una parte fundamental de la solución.
Referencias y Notas
C. P. O’Brien and A. T. McLellan, Lancet 347, 237 (1996).
A. T. McLellan et al., in Treating Drug Abusers Effectively, J. A. Egertson et al., Eds. (Blackwell, Malden, MA, 1997), pp. 7–40.
R. Booth et al., Drug Alcohol Depend. 42, 11 (1996); H. M. Colon et al., AIDS Educ. Prev. 7, 195 (1995); R. C. Stephens et al., in Handbook on Risk of AIDS, B. S. Brown and G. M. Beschner, Eds. (Greenwood, Westport, CT 1993), pp. 519–556; W. W. Wiebel et al., J. Acquired Immune Defic. Syndr. 12, 282 (1996).
American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders, (American Psychiatric Association Press, Washington, DC, ed. 4, 1994); Institute of Medicine, Pathways of Addiction (National Academy Press, Washington, DC, 1996).
G. F. Koob, Trends Pharmacol. Sci. 13, 177 (1992); G. F. Koob et al., Semin. Neurosci. 6, 221 (1994).
S. E. Hyman, Neuron 16, 901 (1996); E. J. Nestler, ibid., p. 897; W. P. Melega et al., Behav. Brain Res. 84, 259 (1997); J. Ortiz et al., Synapse 21, 289 (1995); N. D. Volkow et al., Am. J. Psychiatry 147, 719 (1990).
E. J. Nestler et al., Mol. Psychiatry 1, 190 (1996); D. W. Self and E. J. Nestler, Annu. Rev. Neurosci. 18, 463 (1995).
E. J. Nestler, J. Neurosci. 12, 2439 (1992); T. E. Robinson and K. C. Berridge, Brain Res. Rev. 18, 247 (1993); R. Z. Terwilliger et al., Brain Res. 548, 100 (1991).
G. F. Koob, Neuron 16, 893 (1996).
A. I. Leshner, Hospital Practice: A Special Report (McGraw-Hill, Minneapolis, MN, 1997).
El estado de adicción, tanto la condición clínica como el estado del cerebro, es cualitativamente diferente de los efectos de grandes cantidades de drogas. El individuo, una vez adicto, se ha mudado de un estado donde el uso de drogas es voluntario y controlado a uno donde el deseo, la búsqueda y el uso de drogas ya no están bajo el mismo tipo de control voluntario, y estos cambios reflejan cambios en la función cerebral. No se conocen los mecanismos exactos involucrados. Por ejemplo, no está claro si ese cambio en el estado refleja un cambio relativamente precipitado en un mecanismo único o múltiples mecanismos que actúan en concierto, o si el cambio a la adicción representa la suma de neuroadaptaciones más graduales. Además, hay diferencias individuales en la vulnerabilidad a volverse adictos y la velocidad de volverse adictos. Para algunos individuos, el cambio metafórico se mueve rápidamente, mientras que para otros los cambios ocurren muy gradualmente. (6–10).
L. B. Baxter et al., Semin. Clin. Neuropsychiatry 1, 32 (1996).
A. R. Childress et al., Natl. Inst. Drug Abuse Res. Monogr. 84, 25 (1988); D. C. Daley and G. A. Marlatt, in Substance Abuse: A Comprehensive Textbook, J. H. Lowinson et al., Eds. ( Williams & Wilkins, Baltimore, ed. 3, 1997), pp. 458–467; C. P. O’Brien, Pharmacol. Rev. 27, 535 (1975); C. P. O’Brien et al., Addict. Behav. 15, 355 (1990); S. Grant et al., Proc. Natl. Acad. Sci. U.S.A. 93, 12040 (1996).
J. A. Inciardi et al., J. Drug Issues 27, 261 (1997); H. K. Wexler and D. S. Lipton, in Drug Treatment and Criminal Justice, J. A. Inciardi, Ed. (Sage, Newbury Park, CA, 1993), pp. 261–278.
[1] El área tegmental ventral de Tsai (ATV) es un grupo de neuronas localizadas cerca de la línea media del piso del mesencéfalo. El ATV es el punto de origen donde se encuentran los cuerpos de las células dopaminérgicas del sistema dopaminérgico mesocorticolímbico, y se encuentra ampliamente implicado en el sistema de recompensa natural del cerebro, el mismo que actúa en numerosas adicciones. Es importante en la cognición, motivación, orgasmo, dependencia a las drogas, emociones intensas relacionadas con el amor, y varios desórdenes psiquiátricos. El ATV contiene neuronas que se proyectan hacia numerosas áreas del cerebro, desde la corteza prefrontal (CPF) hasta el tallo cerebral pasando por numerosas regiones entre estas dos. [@kma]
[2] El núcleo accumbens (TA) es un grupo de neuronas del encéfalo, ubicadas donde el núcleo caudado y la porción anterior del putamen confluyen lateralmente con respecto al septum pellucidum. A este núcleo se atribuye una función importante en el placer incluyendo la risa y la recompensa, así como el miedo, la agresión, la adicción y el efecto placebo por lo que se encuentra implicado en el circuito de premio-recompensa. [@kma]
Resumen: La adicción a las drogas es un trastorno crónico recurrente que se ha caracterizado por (1) la compulsión de buscar y tomar la droga, (2) la pérdida de control para limitar el consumo y (3) la aparición de un estado emocional negativo (p. Ej., Disforia, ansiedad, irritabilidad) que refleja un síndrome de abstinencia motivacional cuando se impide el acceso a la sustancia. La adicción a las drogas se ha conceptualizado como un trastorno que involucra elementos tanto de impulsividad como de compulsividad que producen un ciclo compuesto de adicción que comprende tres etapas: «embriaguez/intoxicación», «abstinencia/afecto negativo» y «preocupación/anticipación» (craving). Los estudios de imágenes en animales y humanos han revelado circuitos discretos que median las tres etapas del ciclo de adicción con elementos clave del área tegmental ventral y el estriado ventral como punto focal para la etapa de embriaguez / intoxicación, un papel clave para la amígdala extendida en la etapa de abstinencia / efecto negativo, y un papel clave en la etapa de preocupación / anticipación para una red ampliamente distribuida que incluya la corteza orbitofrontal – cuerpo estriado dorsal, corteza prefrontal, amígdala basolateral, hipocampo e ínsula involucrada en el deseo y la circunvolución cingulada, dorsolateral prefrontal y cortezas frontales inferiores en el control inhibitorio interrumpido. La transición a la adicción implica la neuroplasticidad en todas estas estructuras que pueden comenzar con cambios en el sistema de dopamina mesolímbico y una cascada de neuroadaptaciones desde el estriado ventral al estriado dorsal y la corteza orbitofrontal y, eventualmente, la desregulación de la corteza prefrontal, el giro del cingulado y la amígdala amplificada.
Marco conceptual
Definiciones de adicción: consumo de drogas, abuso y dependencia, ciclo de adicción
La adicción a las drogas es un trastorno crónico recurrente que se ha caracterizado por (1) la compulsión de buscar y tomar la droga, (2) la pérdida de control para limitar el consumo y (3) la aparición de un estado emocional negativo (p. Ej., disforia, ansiedad, irritabilidad) que refleja un síndrome de abstinencia motivacional cuando se impide el acceso a la droga (definido como dependencia de sustancias por el Manual diagnóstico y estadístico de trastornos mentales [DSM] de la American Psychiatric Association; Koob y Le Moal, 1997; Tabla 1). El uso ocasional pero limitado de una sustancia tóxica es clínicamente distinto al uso intensivo de drogas, la pérdida de control sobre la limitación de la ingesta y la aparición de la búsqueda compulsiva crónica de drogas que caracteriza a la adicción. La naturaleza crítica de la distinción entre uso de drogas, abuso y dependencia se ha visto iluminada por datos que muestran que aproximadamente el 15.6% (29 millones) de la población adulta de los EE. UU. pasarán a participar en el consumo de drogas no médicas o ilícitas en algún momento de sus vidas, con aproximadamente el 2,9% (5,4 millones) de dependencia de sustancias de drogas ilícitas (Grant y Dawson, 1998; Grant et al, 2004). Para el alcohol, el 51% (120 millones) de las personas mayores de 12 años son usuarios actuales, y de estos usuarios actuales, el 7,7% (18 millones) cumple con los criterios de abuso de sustancias o dependencia del alcohol. Para la nicotina, en 2007, aproximadamente el 28.6% (70.9 millones) de los estadounidenses mayores de 12 años son usuarios actuales (el mes pasado) de un producto de tabaco, y de estos usuarios actuales, el 24.2% (60.1 millones) son fumadores actuales de cigarrillos; 5.4% (13.3 millones) de cigarros puros; El 3,2% (8,1 millones) consumía tabaco sin humo; y 0,8% (2,0 millones) de tabaco fumado en pipas (Administración de Servicios de Abuso de Sustancias y Salud Mental, 2008).
Definiciones. Tabla 1
Aunque gran parte del estudio inicial de la neurobiología de la adicción a las drogas se centró en el impacto agudo de las drogas de abuso (análogo a la comparación de no usar drogas), ahora se está cambiando a la administración crónica y a los cambios neuroadaptativos agudos y a largo plazo. En el cerebro que dan lugar a recaída. El propósito de la investigación neurobiológica actual sobre el uso indebido de drogas es comprender los mecanismos genéticos/epigenéticos, celulares y moleculares que median la transición del uso ocasional y controlado de drogas a la pérdida de control de la conducta en la búsqueda de drogas y al consumo de drogas, y hasta la recaída crónica después de la abstinencia prolongada que es un sello de la adicción.
Un marco psiquiátrico motivacional que proporciona fuentes de refuerzo positivo y negativo para el consumo de drogas es la conceptualización de que la adicción a las drogas tiene aspectos tanto de los trastornos del control de impulsos como de los trastornos compulsivos (Tabla 1). Los trastornos de control de impulsos se caracterizan por un aumento de la sensación de tensión o excitación antes de cometer un acto impulsivo y de placer, gratificación o alivio en el momento de cometer el acto. Los trastornos del control de impulso se asocian en gran medida con mecanismos de refuerzo positivos (American Psychiatric Association, 1994). En contraste, los trastornos compulsivos se caracterizan por la ansiedad y el estrés antes de cometer un comportamiento compulsivo repetitivo y el alivio del estrés al realizar el comportamiento compulsivo. Los trastornos compulsivos se asocian en gran medida con mecanismos de refuerzo negativos y automaticidad.
Al colapsar los ciclos de impulsividad y compulsividad se obtiene un ciclo de adicción compuesto constituido de tres etapas: embriaguez/intoxicación, abstinencia/afecto negativo, preocupación/anticipación, en el que la impulsividad a menudo domina en las primeras etapas y la impulsividad combinada con la compulsividad domina en las etapas posteriores. A medida que un individuo pasa de la impulsividad a la compulsividad, se produce un cambio desde el refuerzo positivo, que conduce el comportamiento motivado, al refuerzo negativo y la automaticidad que impulsa el comportamiento motivado (Koob, 2004; Tabla 1). Estas tres etapas se conceptualizan como interacción entre ellas, se vuelven más intensas y, en última instancia, conducen al estado patológico conocido como adicción. (Koob y Le Moal, 1997; Tabla 2). La transición del uso ocasional de drogas a la adicción implica la neuroplasticidad en todos estos elementos y puede comenzar con el uso inicial de drogas en individuos vulnerables o en períodos de desarrollo particularmente vulnerables (por ejemplo, la adolescencia; Koob et al, 2008b). La presente revisión se centra en los neurocircuitos cerebrales que participan en cada etapa del ciclo de la adicción, cómo cambia con el aumento del compromiso con las drogas de abuso y cómo interactúa para producir el estado patológico conocido como adicción.
Fuentes de refuerzo: Motivación, proceso opositor, énfasis de incentivo.
Los cambios en la motivación para las drogas y las recompensas naturales son un componente clave de la adicción (Tabla 1). El trabajo inicial de Wikler (1952) hizo hincapié en la función de los cambios en los estados de unidad asociados con la dependencia (en este documento se denomina adicción). Los sujetos describieron los cambios de abstinencia como un «hambre» o necesidad primaria y los efectos de la morfina en un estado tal como «saciedad» o gratificación de la necesidad primaria (Wikler, 1952). Aunque Wikler argumentó que el refuerzo positivo se mantuvo incluso en sujetos muy dependientes (por ejemplo, la emoción de la inyección de opioides intravenosos), la adicción produjo una nueva fuente de gratificación, la del refuerzo negativo (Tabla 1).
El concepto de motivación estaba vinculado indisolublemente con los estados hedónicos, afectivos o emocionales en la transición a la adicción por parte de la teoría de la motivación del proceso opuesto de Salomón. Solomon y Corbit (1974) postularon que los estados hedónicos, afectivos o emocionales, una vez iniciados, son modulados automáticamente por el sistema nervioso central con mecanismos que reducen la intensidad de los sentimientos hedónicos. Las respuestas hedónicas positivas en el uso de drogas ocurren poco después de la presentación de un estímulo, se correlacionan estrechamente con la intensidad, la calidad y la duración del reforzador, y muestran tolerancia y abstinencia afectiva o hedónica (abstinencia). Por el contrario, las respuestas hedónicas negativas siguen a las respuestas hedónicas positivas, tienen un inicio lento, se acumulan lentamente hasta llegar a una asíntota, y decaen lentamente con la exposición repetida. El papel de los procesos del oponente comienza temprano en el consumo de drogas, refleja los cambios en la recompensa cerebral y los sistemas de estrés, y luego forma una de las principales motivaciones para la compulsividad en la toma de drogas en forma de síndrome de abstinencia motivacional.
En esta formulación, la manifestación de un síndrome de abstinencia después de la eliminación de la administración crónica de drogas, ya sea aguda o prolongada, se define en términos de los aspectos motivacionales de la dependencia, como la aparición de un estado emocional negativo (p. Ej., disforia, ansiedad, irritabilidad) cuando se impide el acceso a la droga (Koob y Le Moal, 2001), en lugar de los signos físicos de dependencia, que suelen ser de corta duración. De hecho, algunos han argumentado que el desarrollo de tal estado afectivo negativo puede definir la dependencia relativa a la adicción (Russell, 1976; Baker et al, 1987) y que dicho estado afectivo negativo contribuye a la compulsividad a través de mecanismos de refuerzo negativos (Koob y Le Moal, 2005).
Otra conceptualización de los cambios motivacionales asociados con la adicción se deriva del trabajo inicial sobre el refuerzo condicionado, la motivación de incentivos, la sensibilización del comportamiento y el aprendizaje desadaptativo de estímulo-respuesta, todos los cuales se incluyen en la conceptualización motivacional de la prominencia de incentivos. Se presume que las drogas usurpan los sistemas en el cerebro que se aplican para dirigir a los animales a estímulos con relevancia para la preservación de la especie. La hipótesis de la importancia de los incentivos tiene un valor heurístico significativo como elemento común de la adicción a las drogas porque limita el enfoque hacia la búsqueda de drogas a expensas de las recompensas naturales. La observación clínica de que las personas con trastornos por el uso de sustancias tienen un enfoque inusual en la búsqueda de drogas y la exclusión de recompensas naturales se ajusta a la vista de incentivo destacable.
El aumento en la importancia de los incentivos producidos por las drogas psicoestimulantes tiene sus raíces tempranas en la facilitación del refuerzo condicionado y la búsqueda de drogas (Robbins, 1976; Hill, 1970). Aquí, la búsqueda de drogas está controlada por una sucesión de estímulos discriminativos asociados a las drogas que también pueden funcionar como reforzadores condicionados cuando se presentan como consecuencia de respuestas instrumentales (Everitt et al, 2008). Muchos han argumentado que, mediante el aprendizaje asociativo, el estado mejorado de la atención de relevancia se orienta específicamente hacia los estímulos relacionados con las drogas, lo que lleva a un aumento de la compulsión por buscar y tomar drogas (Hyman et al, 2006; Kalivas y Volkow, 2005). La activación subyacente de las estructuras neuronales involucradas en el mantenimiento del estado de incentivo sobresaliente persiste, haciendo que los adictos sean vulnerables a una recaída a largo plazo.
Otra visión de la importancia de los incentivos involucra la sensibilización conductual, que generalmente se mide como respuestas locomotoras aumentadas a la administración repetida de una droga. El paradigma de sensibilización conductual ha proporcionado un gran impulso para explorar no solo el neurocircuito de la adicción, sino también un modelo de la neuroplasticidad que puede ocurrir durante la transición del consumo de drogas a la adicción. Aquí, se presume que un cambio en un estado de atención de incentivo, descrito como «querer» vinculado al uso compulsivo, en oposición al «agrado» vinculado a las respuestas hedónicas, se incrementó progresivamente por la exposición repetida a drogas de abuso (Robinson y Berridge, 1993).
Transición a la adicción: patrones de consumo de drogas, modelos animales
Diferentes drogas producen diferentes patrones de neuroadaptaciones con exposiciones crónicas a drogas. Por ejemplo, los sujetos adictos a los opioides cumplen con la mayoría de los criterios de DSM para la adicción, que incluyen tolerancia dramática y abstinencia (síntomas clásicos asociados con la dependencia física) y la mayoría de los síntomas asociados con la abstinencia motivacional. Un patrón de consumo de drogas por vía intravenosa o fumada evoluciona, incluye la intoxicación, la tolerancia, el aumento de la ingesta y la disforia profunda, las molestias físicas y los signos somáticos de abstinencia durante la misma. Se desarrolla una intensa preocupación por la obtención de opioides (ansia) que a menudo precede a los signos somáticos de abstinencia y está relacionada no solo con los estímulos asociados con la obtención de la droga, sino también con los estímulos asociados con la abstinencia y el estado motivacional aversivo. Se desarrolla un patrón en el que se debe obtener la droga para evitar la disforia severa y el malestar de la abstinencia. Otras drogas de abuso siguen un patrón similar, pero pueden involucrar más la etapa de atracón/intoxicación (psicoestimulantes) o menos borrachera/ intoxicación y más etapas de abstinencia/afecto negativo y de preocupación/anticipación (nicotina y cannabinoides).
Gran parte de los recientes avances en la comprensión de la neurobiología de la adicción ha derivado del estudio de modelos animales de la adicción a drogas específicas, tales como estimulantes, opioides, alcohol, nicotina, y Δ 9 -tetrahidrocannabinol (Δ 9 -THC). Aunque ningún modelo animal de adicción emula completamente la condición humana, los modelos animales permiten la investigación de elementos específicos del proceso de adicción a las drogas. Dichos elementos pueden definirse por modelos de diferentes etapas del ciclo de adicción (ver más arriba; Tabla 2).
Un aumento progresivo en la frecuencia e intensidad del uso de drogas es uno de los principales fenómenos de comportamiento que caracterizan el desarrollo de la adicción y tiene validez con los criterios del DSM: «La sustancia a menudo se toma en cantidades más grandes y durante un período más prolongado de lo previsto» (Asociación Americana de Psiquiatría, 1994). Se han utilizado dos modelos animales, uno que involucra una droga administrado por un experimentador y otro que incluye una droga autoadministrada, para explorar los efectos de la administración repetida de la droga sobre la neuroplasticidad en los neurocircuitos identificados anteriormente. La sensibilización conductual generalmente involucró la administración repetida por parte del experimentador de una droga, generalmente un psicoestimulante, en un contexto ambiental específico y la medida dependiente fue usualmente la actividad locomotora. Aquí, los animales que recibieron el fármaco mostraron un aumento mucho más dramático en la actividad locomotora a una dosis de desafío de la droga (sensibilización) que los controles que habían recibido solo medidas repetidas de inyecciones. En los modelos animales de acceso prolongado a la autoadministración de drogas se puede encontrar un marco, quizás con más validez nominal con el cual modelar la transición del uso de drogas a la adicción a las drogas. En este caso, al utilizar la autoadministración de medicamentos por vía intravenosa, el acceso prolongado a los medicamentos se asocia con un aumento en el consumo a lo largo de los días (Koob, 2009a). Este aumento de la autoadministración también se ha observado con el alcohol en el que las ratas beben en exceso durante la abstinencia aguda y prolongada de la inducción de la dependencia, ya sea mediante la dieta líquida crónica o la exposición crónica al vapor (Gilpin y Koob, 2008). Los animales dependientes del alcohol obtienen de manera confiable los niveles de alcohol en la sangre en el rango de 100 a 150 mg, que son equivalentes a los niveles abusados por los consumidores de alcohol moderados a pesados. Se han observado cambios en el refuerzo y los efectos de incentivo de la droga luego del acceso extendido y la inducción de la dependencia, e incluyen una mayor respuesta progresiva (Koob, 2009a), una mayor reincorporación inducida por la droga después de la extinción, una menor latencia al tiempo objetivo en un modelo de pasarela por recompensa de drogas (Deroche-Gamonet et al, 2004 ), y mayor resistencia al castigo en el cual el animal sufrirá un mayor castigo aversivo para obtener la droga (Vanderschuren y Everitt, 2004). Si el consumo mejorado de drogas con acceso extendido refleja una sensibilización de la recompensa (o de una motivación de incentivo) o un estado de déficit de recompensa, o ambos, sigue en discusión (Vezina, 2004).
Neurocircuitos de la adicción: evidencia neuropsicofarmacológica de estudios de animales
Etapa de embriaguez / intoxicación
Nuestra comprensión de los sustratos neurobiológicos para los efectos de refuerzo de las drogas de abuso se puede remontar a un trabajo temprano en la identificación de un sistema de recompensa en el cerebro con el descubrimiento de la recompensa de estimulación cerebral eléctrica o autoestimulación intracraneal por Olds y Milner (1954). La recompensa de estimulación cerebral involucra neurocirugía extensa en el cerebro, pero los sitios más sensibles definidos por los umbrales más bajos involucran la trayectoria del haz del cerebro anterior que conecta el área ventral tegmental (VTA) con el cerebro anterior basal (Olds y Milner, 1954). Todas las drogas de abuso, cuando se administran de forma aguda, disminuyen los umbrales de recompensa de estimulación cerebral (es decir, mayor recompensa; Kornetsky y Esposito, 1979) y cuando se administran crónicamente, aumentan los umbrales de recompensa durante la abstinencia (es decir, disminución de la recompensa; consulte a continuación). Aunque inicialmente se hizo mucho hincapié en el papel de los sistemas de monoaminas ascendentes en el paquete del cerebro anterior medial en la recompensa, primero la norepinefrina (Stein, 1962) y luego la dopamina (Crow, 1973; Wise, 1978), otros sistemas no pataminérgicos en el paquete medial del cerebro. claramente tienen un papel clave en la mediación de la recompensa de estimulación cerebral (Hernández y otros, 2006). De hecho, mucho trabajo sugiere que la activación del sistema de dopamina en el cerebro medio tiene múltiples funciones para incentivar la atención a los estímulos en el ambiente (Robinson y Berridge, 1993) para promover el desempeño de la conducta dirigida a objetivos (Salamone et al, 2007) o activación en general (Le Moal y Simon, 1991). Más recientemente, se ha planteado la hipótesis de que el curso temporal de la señalización de dopamina es un factor clave, ya que el curso temporal más rápido tiene un papel preferencial en la recompensa y la valoración de los resultados de comportamiento previstos y la activación constante de la liberación de dopamina tiene un papel preferencial proporcionando un efecto habilitador en sistemas específicos relacionados con el comportamiento (Schultz, 2007). El trabajo en el dominio de los efectos de refuerzo agudo de las drogas de abuso apoya esta hipótesis en la que el sistema de dopamina mesolímbico es crítico para los efectos de recompensa aguda de las drogas psicoestimulantes, pero tiene una función más habilitadora para todas las drogas de abuso.
Se sabe desde hace mucho tiempo que las propiedades de recompensa aguda de las drogas psicoestimulantes dependen de la activación del sistema de dopamina mesolímbica, pero la activación de este sistema no es necesariamente crítica para los efectos de refuerzo agudo de otras drogas de abuso (Koob, 1992; Nestler, 2005; Hnasko et al, 2005). Las lesiones selectivas de neurotoxina del sistema de dopamina mesocorticolímbica bloquean los efectos de refuerzo de la cocaína y la D- anfetamina (McGregor y Roberts, 1993). En contraste, las lesiones neuroquímicas específicas de dopamina en el núcleo accumbens con 6-hidroxidopamina no bloquearon la autoadministración de heroína o etanol, apoyando esta hipótesis (Koob y Le Moal, 2006).
Utilizando la técnica de autoadministración intracraneal (Tabla 1) y el acondicionamiento intracraneal del lugar (Tabla 1), se ha demostrado que los opioides y el alcohol son autoadministrados directamente en el VTA. Los opioides también producen preferencia de lugar condicionado cuando se inyectan en el VTA. Los opioides, la fenciclidina y los psicoestimulantes se administran directamente en el núcleo accumbens, y los psicoestimulantes producen una preferencia de lugar condicionado cuando se inyectan en el núcleo accumbens. La cocaína y la fenciclidina se administran directamente en la corteza frontal (McBride et al, 1999). El sistema de dopamina mesolímbico se activa por la administración aguda de opioides, etanol, nicotina, y Δ 9 -THC (Di Chiara y Imperato, 1988).
La autoadministración de nicotina intravenosa está bloqueada por lesiones específicas de neurotoxina del sistema de dopamina mesocorticolímbica, y se ha planteado la hipótesis de que la acción neurofarmacológica se debe a la activación del receptor nicotínico de la dopamina principalmente en el VTA y también presinápticamente en el núcleo accumbens (Watkins et al., 2000). Sin embargo, la recompensa de nicotina medida por la preferencia de lugar condicionada parece ser independiente del sistema de dopamina mesocorticolímbica (Laviolette et al, 2002). Otros sustratos implicados en la recompensa de la nicotina incluyen entradas colinérgicas al núcleo pedunculopontino (Yeomans y Baptista, 1997). En el VTA, activación de la β.2 subunidades de receptores nicotínicos parecen ser críticos para la activación de nicotina de las neuronas de dopamina (Mameli-Engvall et al, 2006). Los estudios neurofarmacológicos sobre los cannabinoides han implicado mecanismos tanto de cannabinoides como de opioides. Los antagonistas opioides y cannabinoides de CB 1 bloquean la autoadministración intravenosa de Δ9-THC en monos ardilla (Justinova et al, 2003). De manera similar a otras drogas de abuso, la administración de Δ 9 -THC activa la liberación de dopamina en la cáscara del núcleo accumbens (Tanda et al, 1997).
Por lo tanto, todas las drogas de abuso activan el sistema de dopamina mesolímbico, pero mucha evidencia sugiere que el refuerzo independiente de la dopamina se produce a nivel del núcleo accumbens, lo que sugiere múltiples aportes a la activación de los circuitos de refuerzo crítico en estas regiones del cerebro (Koob, 1992; Nestler, 2005).
El núcleo central de la amígdala (CeA) también tiene una función clave en las acciones de refuerzo agudas de las drogas de abuso. Las microinyecciones de los antagonistas del receptor D 1 de dopamina en el CeA bloquean la autoadministración de cocaína (Caine et al, 1995; McGregor y Roberts, 1993). El sitio más sensible para el ácido γ- aminobutírico (GABA) y el antagonismo opioide de la autoadministración oral de alcohol en ratas no dependientes fue el CeA (Hyytia y Koob, 1995; Heyser et al, 1999). Las lesiones del CeA bloquean la autoadministración oral de alcohol (Moller et al, 1997). Los antagonistas de la serotonina-3 inyectados en el CeA bloquean la autoadministración oral de etanol en ratas no dependientes, un efecto hipotético que posiblemente involucre la capacidad de los antagonistas del receptor de la serotonina-3 para bloquear la liberación de dopamina inducida por fármacos (Dyr y Kostowski, 1995).
Una salida importante del núcleo accumbens es el pálido ventral / sustancia innominada. Consistentes con el núcleo accumbens como un sustrato clave para la recompensa de la droga, las lesiones del pálido ventral son particularmente efectivas para bloquear la motivación para trabajar con cocaína intravenosa y heroína intravenosa (Hubner y Koob, 1990; Robledo y Koob, 1993). Además, el bloqueo de los receptores de dopamina y GABA A en el pálido ventral bloquea los efectos de refuerzo del alcohol (Meléndez et al, 2004; June et al, 2003). Por lo tanto, los elementos del pálido ventral pueden no solo ser críticos para el procesamiento posterior de la señal de recompensa de la droga, sino que también pueden ser modulados directamente por las drogas de abuso.
El cuerpo estriado dorsal no parece tener un papel importante en los efectos de refuerzo agudo del abuso de drogas, pero parece ser reclutado durante el desarrollo de la búsqueda compulsiva de drogas (Everett et al, 2008). Las lesiones de 6-hidroxidopamina del estriado dorsal no bloquean la actividad locomotora inducida por la cocaína o la autoadministración de la cocaína (Roberts, 1992), pero sí bloquean el comportamiento estereotipado inducido por la anfetamina (Kelly y Iversen, 1976; Creese e Iversen, 1974). Usando un programa de segundo orden (Tabla 1), las lesiones del núcleo accumbens y la amígdala basolateral impidieron la adquisición de cocaína (Whitelaw et al, 1996). De manera similar, cuando el núcleo del núcleo accumbens se lesionó de manera selectiva en un lado del cerebro y se combinó con el bloqueo del receptor de dopamina en el estriado dorsal contralateral, no se observó ningún efecto en los animales inmediatamente después de la adquisición, pero se observó una disminución considerable de la búsqueda de cocaína en ratas con estables. respondiendo en un horario de segundo orden (Belin y Everitt, 2008). Estos resultados sugieren que el cuerpo estriado dorsal puede tener un papel menor en los efectos de refuerzo agudo de los fármacos psicoestimulantes, pero un papel clave en la transición al uso compulsivo (Everitt et al, 2008).
Los datos con ratones knockout también proporcionan información clave sobre el papel de la dopamina en los efectos gratificantes de las drogas de abuso. Los ratones alterados genéticamente homocigotos con una falta del receptor de dopamina D 1 no se autoadministran cocaína (Caine et al, 2007). Aunque el informe inicial de que los ratones knockout para el transportador de dopamina (DAT) continuaban autoadministrándose cocaína ( Rocha et al, 1998 ) cuestionó la función de la DAT en los efectos de refuerzo de la cocaína, un estudio reciente mostró que los animales transgénicos que expresaban DAT no se unían La cocaína, pero que funcionaba como portador de recaptación de dopamina, no mostró la recompensa de cocaína medida por la preferencia de lugar condicionado ( Chen et al, 2006a). Estos resultados apoyan la hipótesis de un papel crucial de la DAT en los efectos de refuerzo de la cocaína.
Sobre la base de esta síntesis, se propuso un circuito neurobiológico temprano para la recompensa del fármaco (Koob, 1992) que se ha elaborado y ampliado (Koob y Nestler, 1997; Figura 1). El punto de partida para el circuito de recompensa fue el haz del prosencéfalo medial, compuesto por fibras mielinizadas que conectan bidireccionalmente el tubérculo olfativo y el núcleo accumbens con el hipotálamo y la VTA (Nauta y Haymaker, 1969) e incluyen las vías de monoamina ascendentes como el sistema de dopamina mesocorticolímbico.
Figura 1 Sección sagital a través de un cerebro de roedor representativo que ilustra las vías y los sistemas de receptores implicados en las acciones de refuerzo agudas de las drogas de abuso. La cocaína y las anfetaminas activan la liberación de dopamina en el núcleo accumbens y la amígdala a través de acciones directas en los terminales de dopamina. Los opioides activan los receptores opioides en el VTA, el núcleo accumbens y la amígdala a través de acciones directas o indirectas a través de interneuronas. Los opioides facilitan la liberación de dopamina en el núcleo accumbens mediante una acción ya sea en el VTA o en el núcleo accumbens, pero también tienen la hipótesis de que activan elementos independientes del sistema de dopamina. El alcohol activa el ácido γ- aminobutírico-A (GABA A) los receptores o la liberación de GABA en el VTA, el núcleo accumbens y la amígdala, ya sea por acciones directas en el receptor de GABA A o por la liberación indirecta de GABA. Se presume que el alcohol facilita la liberación de péptidos opioides en el VTA, el núcleo accumbens y el núcleo central de la amígdala. El alcohol facilita la liberación de dopamina en el núcleo accumbens a través de una acción en el VTA o el núcleo accumbens. La nicotina activa los receptores nicotínicos de acetilcolina en el VTA, el núcleo accumbens y la amígdala, ya sea directa o indirectamente, a través de acciones sobre las interneuronas. Los cannabinoides activan el cannabinoide CB 1Receptores en el VTA, núcleo accumbens y amígdala. Los cannabinoides facilitan la liberación de dopamina en el núcleo accumbens a través de un mecanismo desconocido en el VTA o el núcleo accumbens. Las flechas azules representan las interacciones dentro del sistema extendido de la amígdala con la hipótesis de que tienen una función clave en el refuerzo de fármacos. El haz del cerebro anterior medial representa proyecciones ascendentes y descendentes entre el cerebro anterior ventral (núcleo accumbens, tubérculo olfatorio, área septal) y el cerebro medio ventral (VTA) (no se muestra en la figura para mayor claridad). CA, comisura anterior; AMG, amígdala; ARC, núcleo arqueado; BNST, núcleo del lecho de la estría terminal; Cer, cerebelo; PC, caudado-putamen; DMT, tálamo dorsomedial; FC, corteza frontal; Hipopótamo, hipocampo; IF, colículo inferior; LC, locus coeruleus; LH, hipotálamo lateral; N Acc., núcleo accumbens; OT, tracto olfativo; PAG, gris periacueductal; RPn, núcleo pontino reticular; SC, colículo superior; SNr, substantia nigra pars reticulata; VP, pálido ventral; VTA, área tegmental ventral (tomada con permiso de Koob, 2005 ).
Se planteó la hipótesis de que la acción inicial de recompensa de drogas dependía de la liberación de dopamina en el núcleo accumbens para la cocaína, la anfetamina y la nicotina; la activación del receptor de péptido opioide en el VTA (activación de dopamina) y el núcleo accumbens (independiente de la activación de dopamina) para los opiáceos; y sistemas GABA A en el núcleo accumbens y amígdala para el alcohol. El núcleo accumbens está situado estratégicamente para recibir información límbica importante de la amígdala, la corteza frontal y el hipocampo que podría convertirse en acción motivacional a través de sus conexiones con el sistema motor extrapiramidal. Por lo tanto, se estableció un papel crítico inicial para el núcleo accumbens para los efectos de refuerzo agudo de los fármacos, con un papel de apoyo para el CeA y el pálido ventral (Figuras 1 y2a).
Figura 2 Circuitos neuronales asociados a las tres etapas del ciclo de la adicción. (a) Etapa de embriaguez / intoxicación. El refuerzo de los efectos de los fármacos puede comprometer a los neurotransmisores de recompensa y los mecanismos asociativos en el núcleo accumbens de la cáscara y el núcleo, y luego activar hábitos de estímulo-respuesta que dependen del estriado dorsal. Los dos neurotransmisores principales que median los efectos gratificantes de las drogas de abuso son la dopamina y los péptidos opioides. (b) Retiro / etapa de afectación negativa. El estado emocional negativo de abstinencia puede comprometer la activación de la amígdala extendida. La amígdala extendida se compone de varias estructuras basales del cerebro anterior, incluido el núcleo del lecho de la estría terminal, el núcleo central de la amígdala y, posiblemente, una zona de transición en la porción medial (o capa) del núcleo accumbens. Los neurotransmisores principales en la amígdala extendida con la hipótesis de que tienen una función en el refuerzo negativo son el factor liberador de corticotropina, la norepinefrina y la dinorfina. Las principales proyecciones de la amígdala extendida son para el hipotálamo y el tronco cerebral. (c) Etapa de preocupación / anticipación (ansia). Esta etapa implica el procesamiento de refuerzo condicionado en el BLA y el procesamiento de información contextual por parte del hipocampo. El control ejecutivo depende de la corteza prefrontal e incluye la representación de contingencias, la representación de resultados y su valor y estados subjetivos (es decir, ansia y, presumiblemente, sentimientos) asociados con las drogas. Los efectos subjetivos denominados ansia de drogas en humanos implican la activación en estudios de imagen funcionales de las cortezas y el lóbulo temporal del cingulado orbital y anterior, Incluyendo la amígdala. Un importante neurotransmisor involucrado en la etapa de deseo es el glutamato localizado en las vías desde las regiones frontales y el BLA que se proyecta hacia el estriado ventral. Flechas verdes / azules, proyecciones glutamatérgicas; Flechas naranjas, proyecciones dopaminérgicas; flechas rosas, proyecciones gabergicas; Acb, núcleo accumbens; BLA, amígdala basolateral; VTA, área tegmental ventral; SNc, substantia nigra pars compacta; VGP, globo pálido ventral; DGP, globo pálido dorsal; BNST, núcleo del lecho de la estría terminal; CeA, núcleo central de la amígdala; NE, norepinefrina; CRF, factor liberador de corticotropina; PIT, transferencia instrumental pavloviana (modificada con permiso de Flechas verdes / azules, proyecciones glutamatérgicas; Flechas naranjas, proyecciones dopaminérgicas; flechas rosas, proyecciones gabergicas; Acb, núcleo accumbens; BLA, amígdala basolateral; VTA, área tegmental ventral; SNc, substantia nigra pars compacta; VGP, globo pálido ventral; DGP, globo pálido dorsal; BNST, núcleo del lecho de la estría terminal; CeA, núcleo central de la amígdala; NE, norepinefrina; CRF, factor liberador de corticotropina; PIT, transferencia instrumental pavloviana (modificada con permiso de Flechas verdes / azules, proyecciones glutamatérgicas; Flechas naranjas, proyecciones dopaminérgicas; flechas rosas, proyecciones gabergicas; Acb, núcleo accumbens; BLA, amígdala basolateral; VTA, área tegmental ventral; SNc, substantia nigra pars compacta; VGP, globo pálido ventral; DGP, globo pálido dorsal; BNST, núcleo del lecho de la estría terminal; CeA, núcleo central de la amígdala; NE, norepinefrina; CRF, factor liberador de corticotropina; PIT, transferencia instrumental pavloviana (modificada con permiso de núcleo central de la amígdala; NE, norepinefrina; CRF, factor liberador de corticotropina; PIT, transferencia instrumental pavloviana (modificada con permiso de núcleo central de la amígdala; NE, norepinefrina; CRF, factor liberador de corticotropina; PIT, transferencia instrumental pavloviana (modificada con permiso de Koob et al, 2008a ).
Abstinencia / Etapa de afecto negativo
La entidad neuroanatómica denominada amígdala extendida (Heimer y Alheid, 1991) puede representar un sustrato anatómico común que integra los sistemas de activación-activación cerebral con sistemas de procesamiento hedónicos para producir los estados emocionales negativos que promueven los mecanismos de refuerzo negativos asociados con el desarrollo de la adicción. La amígdala extendida está compuesta por el CeA, el núcleo del lecho de la estría terminal (BNST) y una zona de transición en la subregión medial (concha) del núcleo accumbens (Figura 2b). Cada una de estas regiones tiene similitudes de citoarquitecturas y circuitos (Heimer y Alheid, 1991). La amígdala extendida recibe numerosos aferentes de estructuras límbicas como la amígdala basolateral y el hipocampo y envía efferentes a la parte medial del pálido ventral y una gran proyección al hipotálamo lateral, definiendo así las áreas específicas del cerebro que interactúan con el límbico clásico (emocional) Estructuras con la salida del sistema motor extrapiramidal (Alheid et al, 1995). Durante mucho tiempo se ha planteado la hipótesis de que la amígdala extendida tiene un papel clave no solo en el condicionamiento del miedo (Le Doux, 2000), sino también en el componente emocional del procesamiento del dolor (Neugebauer et al, 2004).
Las neuroadaptaciones internas a la exposición crónica a medicamentos incluyen disminuciones en la función de los sistemas de neurotransmisores en los neurocircuitos implicados en los efectos de refuerzo agudo de las drogas de abuso. Una hipótesis importante es que los sistemas de dopamina están comprometidos en fases cruciales del ciclo de la adicción, como la abstinencia, y conducen a una menor motivación para los estímulos no relacionados con los medicamentos y una mayor sensibilidad a la droga de abuso (Melis et al, 2005; vea los estudios de imágenes cerebrales a continuación). La abstinencia psicoestimulante en humanos se asocia con fatiga, disminución del estado de ánimo y retraso psicomotor, y en animales se asocia con menor motivación para trabajar por recompensas naturales (Barr y Phillips, 1999) y disminución de la actividad locomotora (Pulvirenti y Koob, 1993), efectos de comportamiento que pueden implicar disminución de la función dopaminérgica. Los animales durante la abstinencia de anfetaminas muestran una respuesta disminuida en un programa de proporción progresiva para una solución dulce, y esta respuesta disminuida se revirtió con el agonista parcial de dopamina tergurida (Orsini et al, 2001), lo que sugiere que el tono bajo de dopamina contribuye a los déficits motivacionales asociados con Retiro psicoestimulante. La disminución de la actividad del sistema de dopamina mesolímbica y la disminución de la neurotransmisión serotoninérgica en el núcleo accumbens ocurren durante la retirada aguda de drogas de todas las principales drogas de abuso en estudios con animales (Rossetti y otros, 1992; Weiss y otros, 1992, 1996).
Un segundo componente de la etapa de abstinencia / afecto negativo es una neuroadaptación entre sistemas en la cual diferentes sistemas neuroquímicos involucrados en la modulación del estrés también pueden involucrarse dentro de los neurocircuitos del estrés cerebral y los sistemas aversivos en un intento por superar la presencia crónica de la perturbación. Medicamento para restablecer la función normal a pesar de la presencia del fármaco. Tanto el eje hipotálamo-hipófisis-suprarrenal y el estrés cerebral / sistema aversivo mediado por el factor liberador de corticotropina (CRF) se activan durante la abstinencia de la administración crónica de todas las principales drogas con potencial de abuso, con una respuesta común de una hormona adrenocorticotrópica elevada, corticosterona, y CRF de la amígdala durante la retirada aguda (Koob, 2008; Koob y Kreek, 2007). La abstinencia aguda de todas las drogas de abuso también produce un estado aversivo o similar a la ansiedad en el que la CRF y otros sistemas relacionados con el estrés (incluidas las vías noradrenérgicas) tienen funciones clave.
Los efectos de estímulo aversivo de la abstinencia de la droga se pueden medir utilizando la aversión al lugar (Hand et al, 1988), y la dosis de buprenorfina del agonista parcial del opioide redujo de manera dependiente la aversión al lugar producida por la abstinencia del opioide precipitado. La administración sistémica de un antagonista del receptor de CRF 1 y la administración intracerebral directa de un péptido antagonista de CRF 1 / CRF 2 también disminuyeron las aversiones de lugar inducidas por el retiro de opioides (Stinus et al, 2005; Heinrichs et al, 1995). Los antagonistas noradrenérgicos funcionales administrados directamente en la BNST bloquearon la aversión al lugar inducida por el retiro de opioides, lo que implica la importancia de la estimulación noradrenérgica en las respuestas de estrés que siguen a la abstinencia aguda de drogas (Delfs et al, 2000). De hecho, los medicamentos clásicos utilizados para tratar la abstinencia física en los consumidores de alcohol y los alcohólicos incluyen drogas adrenérgicas α (por ejemplo, clonidina) que inhiben la liberación de noradrenérgicos y disminuyen algunos síntomas de abstinencia de alcohol y heroína.
Otro candidato para los efectos aversivos de la abstinencia de drogas es la dinorfina. Mucha evidencia muestra que la dinorfina aumenta en el núcleo accumbens en respuesta a la activación dopaminérgica y, a su vez, que la actividad excesiva de los sistemas de dinorfina puede disminuir la función dopaminérgica. Los agonistas κ- opioides son aversivos, y la abstinencia de cocaína, opioides y etanol se asocia con un aumento de la dinorfina en el núcleo accumbens y / o amígdala (Koob, 2008). Una excepción es la salvidorina A, que es un agonista κ abusado por los humanos, pero esto puede reflejar sus efectos alucinógenos en lugar de propiedades placenteras (Gonzalez et al, 2006).
Otra respuesta común entre sistemas al retiro agudo y la abstinencia prolongada de todas las principales drogas de abuso es la manifestación de respuestas similares a la ansiedad. Por ejemplo, la abstinencia de la administración repetida de cocaína produce una respuesta de tipo ansiogénico en la prueba de enterramiento defensivo elevado plus, y ambos son revertidos por los antagonistas de CRF. Del mismo modo, la retirada de etanol produce un comportamiento similar a la ansiedad que se invierte mediante la administración intracerebroventricular de CRF 1 / CRF 2 antagonistas peptidérgicas, la administración sistémica de una molécula pequeña CRF 1 antagonista, y la microinyección de un peptidergic CRF 1 / CRF 2 antagonista en la amígdala (Funk et al, 2006; Koob, 2008). Los antagonistas de CRF inyectados intracerebroventricular o sistémicamente también bloquean las respuestas similares a la ansiedad potenciada a los estresores observados durante la abstinencia prolongada de etanol crónico, y los efectos de los antagonistas de CRF se han localizado en el CeA (Koob, 2008). La abstinencia precipitada de la nicotina produce respuestas similares a la ansiedad que también son revertidas por los antagonistas de CRF (Tucci et al, 2003; George et al, 2007).
Por lo tanto, la abstinencia aguda se asocia con cambios dentro del sistema que se reflejan en una disminución de la actividad dopaminérgica en el sistema de dopamina mesolímbica y con el reclutamiento entre sistemas de sistemas de neurotransmisores que transmiten estrés y efectos similares a la ansiedad, como CRF y dinorfina. Otros sistemas de neurotransmisores que se sabe están involucrados en la desregulación emocional de los efectos motivacionales de la abstinencia del fármaco incluyen norepinefrina, sustancia P, vasopresina, neuropéptido Y (NPY), endocannabinoides y nociceptina (Koob, 2008)
Etapa de Preocupación / Anticipación (Craving)
La etapa de preocupación / anticipación o antojo del ciclo de adicción ha sido durante mucho tiempo la hipótesis de ser un elemento clave de la recaída en los seres humanos y define la adicción como un trastorno crónico de recaída. Aunque a menudo está relacionado con la construcción del deseo, el deseo en sí mismo ha sido difícil de medir clínicamente (Tiffany et al, 2000) y a menudo no se correlaciona bien con la recaída. Sin embargo, la etapa del ciclo de adicción en la que el individuo restablece el comportamiento de búsqueda de drogas después de la abstinencia sigue siendo un enfoque desafiante para los mecanismos neurobiológicos y el desarrollo de medicamentos para el tratamiento. Los modelos animales de deseo se pueden dividir en dos dominios: búsqueda de drogas inducida por drogas o estímulos combinados con la toma de drogas, y búsqueda de drogas inducida por un estresante agudo o un estado emocional negativo residual, a menudo un estado de estrés, denominado abstinencia prolongada (Ver Transición a la adicción: patrones de consumo de drogas, sección de modelos animales).
Mucha evidencia de estudios en animales sugiere que el restablecimiento inducido por fármacos se localiza en el córtex prefrontal medial / núcleo accumbens / pálido ventral, mediado por el neurotransmisor glutamato (McFarland y Kalivas, 2001). En contraste, el restablecimiento inducido por el indicio parece involucrar a la amígdala basolateral como un sustrato crítico con un posible mecanismo de avance a través del sistema de corteza prefrontal involucrado en el restablecimiento inducido por fármacos (Everitt y Wolf, 2002; Weiss et al, 2001). La asociación de estímulos previamente neutros combinados con la abstinencia de opioides precipitados (abstinencia condicionada) también depende fundamentalmente de la amígdala basolateral (Schulteis et al, 2000).), y tales estímulos pueden tener un significado motivacional (Kenny et al, 2006). Los cambios neurocircuiticos asociados con el restablecimiento inducido por medicamentos y señales después de la extinción se han vinculado a una vía glutamatérgica desde la corteza prefrontal al núcleo del núcleo accumbens, la proyección de dopamina desde el VTA a la corteza prefrontal medial y la proyección GABA desde el núcleo accumbens al pálido ventral (Kalivas y O’Brien, 2008).
En contraste, el restablecimiento inducido por el estrés de la respuesta relacionada con el fármaco en modelos animales parece depender de la activación de CRF y norepinefrina en elementos de la amígdala extendida (tanto el CeA como el BNST; para revisiones, ver Shaham et al, 2003; Shalev et al, 2002). La abstinencia prolongada, descrita en gran parte en los modelos de dependencia del alcohol, parece implicar sistemas glutamatérgicos y CRF hiperactivos, probablemente en la amígdala extendida, aunque en gran medida esto aún no se ha explorado (de Witte et al, 2005; Valdez et al, 2002).
Los sujetos humanos con adicción a la cocaína muestran un desempeño deficiente en tareas que involucran atención, flexibilidad cognitiva y descuentos de recompensa diferida que están mediados por las cortezas prefrontales medial y orbital, así como también las discapacidades de memoria espacial, verbal y de reconocimiento que está mediada por el hipocampo, y estos déficits pueden predecir malos resultados de tratamiento (Aharonovich et al, 2006; Bolla et al, 2003). Los estudios en animales paralelos del orbitofrontal, la corteza prefrontal y el hipocampo en adicción con modelos animales han comenzado a mostrar algunos de los déficits reflejados en estudios en humanos. La cocaína administrada por el experimentador produjo alteraciones en el aprendizaje de reversión (una tarea frontal orbital) en ratas y monos (Jentsch et al, 2002; Schoenbaum et al, 2004; Calu et al, 2007). Tal vez aún más convincente, los animales permitieron un acceso extendido, pero no limitado, a la cocaína que mostró deficiencias en la memoria de trabajo (una tarea que depende de la corteza prefrontal), una tarea de atención sostenida (una tarea que depende de la corteza prefrontal) y una tarea de reconocimiento de objetos. (una tarea dependiente del hipocampo; Briand et al, 2008a, 2008b; George et al, 2008). En un estudio (Briand et al, 2008a), estos déficits se asociaron con una disminución significativa de la dopamina D 2. El ARNm del receptor en la corteza prefrontal medial y orbital, una observación que también concuerda con los estudios de imágenes en humanos. Por lo tanto, los estudios en animales que utilizan modelos de administración de estimulantes compulsivos están comenzando a mostrar déficits asociados con la adicción a la cocaína humana (ver Estudios en humanos: imágenes y neuropsicofarmacología).
Estudios humanos: imagen y neuropsicofarmacología
Como se señaló anteriormente, la evidencia de estudios preclínicos y clínicos sugiere que la adicción representa neuroadaptaciones secuenciales. Como resultado, una acción impulsiva inicial se vuelve compulsiva y se vuelve (eventualmente) crónica y recurrente. El trabajo de los estudios de imágenes ha proporcionado evidencia de que esta transición implica la reprogramación de circuitos neuronales que procesan (1) la recompensa y la motivación; (2) memoria, condicionamiento y habituación; (3) función ejecutiva y control inhibitorio; (4) interocepción y autoconciencia; y (5) reactividad al estrés. Esta transición está fuertemente influenciada por factores genéticos, de desarrollo y ambientales y sus interacciones dinámicas, que determinarán el curso y la gravedad de la adicción.
Al igual que en las investigaciones preclínicas, ha sido útil distinguir las tres etapas en el curso recurrente de la adicción en humanos (intoxicación, abstinencia y ansia / recaída). Las siguientes secciones describen estas etapas y algunos de los circuitos neuronales relevantes que las subyacen.
Etapa de embriaguez / intoxicación
La mayoría de los casos de adicción se inician por el abuso de sustancias que se buscan debido a sus propiedades hedónicas. Sin embargo, la experimentación con drogas también resulta de los efectos de refuerzo de conformarse con grupos sociales (presión de grupo) con la eventual transferencia de motivación para tomar la droga por sus efectos de refuerzo. Con poca frecuencia, el primer uso de una droga puede estar relacionado con sus propiedades terapéuticas (como los analgésicos opiáceos para el dolor o los estimulantes para el trastorno por déficit de atención con hiperactividad). Como lo demuestran los estudios preclínicos, se acepta ampliamente que un elemento clave de los efectos de refuerzo de los fármacos implica su capacidad para desencadenar grandes aumentos de dopamina extracelular en las regiones límbicas (incluido el núcleo accumbens). Aunque la autoadministración aguda de drogas es un buen modelo animal para la intoxicación por drogas, el uso de modelos animales para evaluar los correlatos subjetivos de los aumentos de dopamina inducidos por drogas es difícil. Los estudios de imágenes cerebrales en humanos han sido fundamentales para demostrar que los aumentos inducidos por drogas en la dopamina en el cuerpo estriado (incluido el cuerpo estriado ventral donde se localiza el núcleo accumbens) están asociados con descriptores subjetivos de recompensa (por ejemplo, placer, subidón, euforia; Volkow et al, 1996b). Además, estos estudios han demostrado que los cambios rápidos de dopamina se asocian con la percepción subjetiva de la recompensa, mientras que los aumentos de dopamina lentos y estables no inducen estas respuestas subjetivas (Grace, 2000; Volkow y Swanson, 2003).
Las propiedades farmacocinéticas de las drogas, que influyen en la velocidad de administración en el cerebro y en la duración de sus acciones, son elementos clave de su potencial de adicción. Las propiedades farmacocinéticas determinan las dosis, las vías de administración y la frecuencia de uso del fármaco dentro de un episodio de atracón dado. Por ejemplo, la comparación de la farmacocinética cerebral de la cocaína y la metanfetamina revela que ambas llegan al cerebro muy rápidamente (aunque la cocaína es algo más rápida que la metanfetamina) pero que la cocaína se elimina del cerebro mucho más rápido que la metanfetamina (Figura 3). Esta diferencia ayuda a explicar por qué se toma cocaína cada 30 a 60 minutos durante un atracón, mientras que la metanfetamina se toma cada dos horas (Fowler et al, 2008). La importancia de la farmacocinética también ayuda a explicar por qué la mayoría de las drogas de abuso (con la excepción del alcohol) se inyectan, fuman o aspiran. Estas rutas permiten un suministro mucho más rápido de la droga al cerebro que cuando se toma por vía oral (Volkow et al, 2000). La farmacocinética también ayuda a explicar por qué los medicamentos estimulantes como el metilfenidato o la anfetamina, que también aumentan la dopamina, no suelen percibirse como reforzantes cuando se toman oralmente según lo prescrito terapéuticamente (Chait, 1994; Volkow et al, 2001b).
Figura 3 Imágenes cerebrales obtenidas en diferentes momentos después de la administración de [ 11 C] D -metanfetamina y de [ 11 C] cocaína (n = 19 para cada medicamento) que muestran planos axiales a un nivel que atraviesa los ganglios basales. Tenga en cuenta la rápida absorción de ambos fármacos en el cerebro y el aclaramiento mucho más lento para la [ 11C] D -metanfetamina que para la [ 11 C] cocaína (tomada con el permiso de Fowler et al, 2008).
Los estudios clínicos también han demostrado que la expectativa de los efectos del fármaco influye significativamente en las respuestas gratificantes a los medicamentos, de modo que la respuesta de activación cerebral y del comportamiento al cerebro tiende a ser más intensa cuando se espera un fármaco gratificante en comparación con cuando la misma sustancia se recibe inesperadamente (Volkow et al, 2003). La dependencia de los efectos gratificantes de la droga en el contexto y la expectativa sugiere la importancia de otros neurotransmisores como el glutamato, que modula la reactividad de las células de dopamina y la liberación de dopamina en el núcleo accumbens, en los efectos gratificantes de las drogas de abuso (Kalivas y Volkow, 2005).
Abstinencia / Etapa de afecto negativo
La respuesta que sigue a la etapa de intoxicación por drogas difiere notablemente entre las drogas y está influenciada por la cronicidad y la frecuencia de su abuso. Para algunas drogas, como los opiáceos, el alcohol y los hipnóticos sedantes, la interrupción de la droga en los usuarios crónicos de drogas puede desencadenar un síndrome de abstinencia física intenso y agudo que, si no se controla adecuadamente y cuando es grave, a veces puede ser fatal. Todas las drogas de abuso están asociadas con un síndrome de abstinencia motivacional caracterizado por disforia, irritabilidad, malestar emocional y trastornos del sueño que persisten incluso después de una abstinencia prolongada. La neurobiología de la abstinencia aguda es distinta de la abstinencia prolongada o motivacional, y ambas contribuyen a la recaída. Se han realizado pocos estudios de imagen durante la retirada aguda. (Wang et al, 1997). De este estudio, no está claro si los resultados reflejan la falta de participación de la dopamina del estriado durante la abstinencia aguda en los consumidores de heroína o la sensibilidad limitada de la tecnología de tomografía por emisión de positrones (PET).
Es probable que los mecanismos que subyacen a la abstinencia aguda sean específicos de la droga y reflejen adaptaciones en los objetivos moleculares de estas drogas. Por ejemplo, durante los primeros días de abstinencia de la cocaína, se produce una mayor sensibilidad del cerebro a los efectos de las drogas que aumentan el GABA que pueden reflejar la regulación a la baja de este neurotransmisor con el uso crónico de cocaína (Volkow y otros, 1998). De manera similar, los estudios de imágenes cerebrales también han revelado disminuciones en los opioides endógenos durante el retiro de la cocaína, lo que puede contribuir a la irritabilidad, el malestar y la disforia que ocurren durante esta fase de retiro motivacional (Zubieta et al, 1996).
Durante la abstinencia prolongada, una vez que los signos y síntomas de la abstinencia aguda han disminuido, los estudios de imagen han documentado una hipofunción en las vías de dopamina, demostrada por disminuciones en la expresión del receptor D 2 y disminuciones en la liberación de dopamina, lo que puede contribuir a la anhedonia (es decir, una menor sensibilidad a estímulos gratificantes) y la amotivación informada por sujetos drogadictos durante la abstinencia prolongada (Volkow y otros, 1997b , 2007; Martinez y otros, 2004, 2005). La disminución de la reactividad de la dopamina a los estímulos de refuerzo también está presente después de la abstinencia prolongada del alcohol cuando la abstinencia física aguda ha disminuido. En contraste con la disminución de la sensibilidad a las recompensas (incluidas las recompensas por medicamentos), los estudios de imagen han informado que, durante la desintoxicación, también se produce una mayor sensibilidad a las señales condicionadas. La abstinencia de fumar, por ejemplo, puede potenciar dramáticamente las respuestas neuronales a las señales relacionadas con el hábito de fumar (McClernon et al, 2009). Estas respuestas condicionadas sostienen el ciclo de abstinencia y recaída que caracteriza los trastornos por uso de sustancias (Childress et al, 1988).
Además, los estudios de imagen que evalúan los marcadores de la función cerebral han demostrado que los drogadictos analizados durante la desintoxicación prolongada muestran evidencias de actividad interrumpida de las regiones frontales, incluidas las regiones prefrontales dorsolaterales, la circunvolución cingulada y la corteza orbitofrontal, que se supone que subyacen a su control inhibitorio deficiente e impulsividad y contribuir a la recaída (consulte la siguiente sección para obtener información).
Etapa de Preocupación / Anticipación (Craving)
La mayor sensibilidad a las señales condicionadas, que incluyen estados emocionales, desencadena la etapa de preocupación / anticipación (deseo) latente, que se caracteriza por un aumento en el deseo por las drogas. De hecho, el estrés es un poderoso desencadenante de la recaída a los comportamientos de consumo de drogas mediante la activación de circuitos cerebrales involucrados en el procesamiento de recompensas y en el sesgo de atención y mnemotécnico para los recordatorios de uso de drogas (Duncan et al, 2007). Este fenómeno de recaída crónica es ampliamente reconocido como uno de los problemas más desafiantes en la lucha contra la adicción a las drogas. Los sujetos adictos pueden regresar a la toma compulsiva de drogas mucho después de experimentar síntomas agudos de abstinencia (Langleben et al, 2008). Se cree que la reorganización gradual de los circuitos de recompensa y memoria, provocada por el abuso crónico de drogas, es crucial para el montaje de estas respuestas. Tanto la dopamina como el glutamato se han identificado en estudios preclínicos que contribuyen a los cambios neuroplásticos asociados con las respuestas condicionadas. Además, los cambios plásticos en los receptores de CRF y glucocorticoides probablemente participan en la sensibilidad aumentada a los factores estresantes. En los seres humanos, la falta de radiotrazadores adecuados para evaluar la neurotransmisión de glutamato y la falta de ligandos para los receptores de IRC o glucocorticoides han limitado los estudios de ansia principalmente al sistema de dopamina.
Dinámicas de los circuitos neurológicos en la transición a la adicción
Los neurocircuitos descritos anteriormente forman la base de la neuroplasticidad asociada con el desarrollo de la adicción. A continuación, se resumen los cambios neuroadaptativos que participan en los circuitos que representan las etapas del ciclo de adicción descrito anteriormente. Se presume que cinco circuitos se involucran en la sucesión, incluyendo (1) sistema de dopamina mesolímbica, (2) estriado ventral, (3) estriado ventral / estriado dorsal / circuitos del tálamo, (4) cortex dorsolateral / circuitos de la corteza frontal inferior / hipocampo, y (5) amígdala extendida (Figura 4). La ponderación relativa y la dirección de estos cambios neuroadaptativos se ilustran en el diagrama del circuito del estado adicto (Figura 5).
Figura 4 Dibujo esquemático que describe los efectos secuenciales y acumulativos de los cambios neuroadaptativos con la hipótesis de contribuir a la neuroplasticidad que promueve la búsqueda compulsiva de drogas. Una neuroadaptación temprana, común a todas las drogas de abuso y observada después de una sola inyección de cocaína, es el aumento de la excitabilidad del sistema de dopamina mesolímbico reflejado en la potenciación a largo plazo dependiente de los cambios en la actividad del glutamato. Posteriormente, la activación de la dopamina contribuye a aumentar la excitabilidad del cuerpo estriado ventral con una actividad glutamatérgica disminuida durante la abstinencia y una actividad glutamatérgica aumentada durante la búsqueda de drogas inducida por el fármaco e inducida por el indicio. La participación de los lazos ventrales estriado-pálido-talámico está supuestamente traducida al estriado dorsal para contribuir a la participación de hábitos y automaticidad que se asemejan a un comportamiento compulsivo. A medida que la compulsividad evoluciona hacia una adicción en toda regla, la pérdida de la función ocurre en los sistemas de la corteza frontal que controlan la función ejecutiva, lo que contribuye a la toma de decisiones deficiente y la ganancia de la función en los sistemas de estrés cerebral, pero contribuye a fomentar la prominencia de las drogas sobre los refuerzos naturales.
Figura 5 Esquema de neurocircuitos que ilustra la combinación de neuroadaptaciones en los circuitos cerebrales para las tres etapas del ciclo de adicción que promueven el comportamiento de búsqueda de drogas en el estado adicto. Tenga en cuenta la activación del cuerpo estriado ventral / cuerpo estriado dorsal / amígdala extendida conducida por señales a través del hipocampo y la amígdala basolateral y el estrés a través de la ínsula. El sistema de la corteza frontal está comprometido, produciendo déficits en la función ejecutiva y contribuyendo a la prominencia de incentivo de las drogas en comparación con los reforzadores naturales. Los sistemas de dopamina están comprometidos y los sistemas de estrés cerebral como CRF se activan para restablecer aún más la prominencia de las drogas y los estímulos relacionados con las drogas en el contexto de un estado disfórico aversivo (modificado con el permiso de Koob et al, 2008a ).
Sistema mesolímbico de dopamina: vías de incentivo a la saliencia, atribución a la saliencia
Una hipótesis importante que guía la neuroplasticidad asociada con la adicción se centra en el sistema de dopamina mesolímbica. La hipótesis es que las drogas de abuso, en particular la cocaína y la anfetamina, aumentan la liberación de dopamina de manera más prolongada y no regulada que los estímulos naturales, lo que produce cambios en la plasticidad sináptica tanto dentro del sistema de dopamina como en las neuronas receptoras de dopamina (Wolf, 2002). Estos cambios, en última instancia, usurpan los mecanismos de aprendizaje normal para hacer que los neurocircuitos se conviertan en asociaciones o en una forma de aprendizaje de hábitos que persiste frente a las consecuencias adversas significativas (un componente de la compulsividad; Everitt y Wolf, 2002; Hyman et al, 2006).
Los modelos animales de sensibilización conductual se han centrado en gran medida en el aumento de los efectos de activación locomotora de los fármacos estimulantes psicomotores en animales con antecedentes de exposición a estimulantes. Tales estudios han revelado una rica neuroplasticidad asociada con los sistemas de dopamina mesolímbicos y su proyección terminal al estriado ventral (donde se encuentra el núcleo accumbens). Las drogas de abuso inducen modificaciones a corto y largo plazo de la activación de las neuronas de dopamina en el VTA (Bonci et al, 2003). Los estudios han demostrado que la activación de ráfagas de neuronas de dopamina en el VTA parece estar correlacionada con una respuesta de orientación a un estímulo sensorial (Freeman et al, 1985). Una sola exposición in vivo a la cocaína o anfetamina induce la potenciación a largo plazo (LTP) de la neurotransmisión excitadora mediada por AMPA en neuronas de dopamina (Ungless et al, 2001). Se ha planteado la hipótesis de que la potenciación de las respuestas sinápticas de AMPA aumenta la incidencia de disparos en ráfaga (Jones y Bonci, 2005). La LTP persistente que duró 3 meses de abstinencia se indujo en el VTA en ratas que autoadministraron cocaína activamente pero no en ratas inyectadas pasivamente (Chen et al, 2008). Se han observado efectos similares de la inducción de la LTP de la transmisión de glutamato en las neuronas de dopamina con morfina y nicotina (Saal et al, 2003).
Sin embargo, la administración repetida más crónica de psicoestimulantes no logró producir la sensibilización de la actividad de dopamina mesolímbica medida por microdiálisis in vivo (Maisonneuve et al, 1995). Además, el acceso extendido a la cocaína no produce sensibilización locomotora (Ben-Shahar et al, 2004) pero sí produce una respuesta de comportamiento estereotipada sensibilizada (Ferrario et al, 2005). Además, los consumidores de cocaína humanos mostraron respuestas de dopamina atenuadas cuando se les aplicó un medicamento estimulante, que es opuesto al predicho por la sensibilización aumentada de la actividad de dopamina mesolímbica (Volkow et al, 1997b; Martinez et al, 2007).
Estriado ventral: vías de atención de incentivo, atribución de saliencia
Otra plasticidad asociada con la sensibilización conductual es una potenciación persistente de las sinapsis excitatorias del núcleo accumbens que se observa después de la exposición repetida al fármaco seguida de un período prolongado libre de fármaco (Kourrich et al, 2007). La administración repetida de cocaína aumenta la neurotransmisión de glutamato solo en ratas que mostraron sensibilización de comportamiento (Pierce et al, 1996). Además, los ratones sensibilizados con cocaína mostraron un aumento de la LTP en los cortes de núcleo accumbens durante la extracción, lo que probablemente refleja un aumento de la actividad de la actividad glutamatérgica (Yao et al, 2004).). 21 días después de la última inyección de cocaína, se observó un aumento de la relación superficie-intracelular de los receptores de glutamato-1 (GluR1), lo que sugiere un desarrollo lento de la redistribución de los receptores de AMPA en la superficie de las neuronas del núcleo accumbens, particularmente en las que carecen de GluR2 Boudreau y Wolf, 2005; Conrad et al, 2008). Los aumentos en los receptores AMPA de la superficie celular dependen de la activación de los receptores de dopamina D 1 y la señalización de la proteína quinasa A posterior (Chao et al, 2002). Funcionalmente, la sobreexpresión de GluR1 en el núcleo accumbens facilitó la extinción de las respuestas en busca de cocaína (Sutton et al, 2003) y el aumento de la estimulación cerebral recompensa los umbrales, lo que refleja una disminución de la recompensa y posiblemente una disminución de la conducta motivada (Todtenkopf et al, 2006). Sin embargo, una sola reexposición a la cocaína durante la abstinencia prolongada produjo depresión sináptica, lo que puede reflejar la mayor liberación de glutamato durante la reexposición de la cocaína (Kourrich et al, 2007). Curiosamente, el aumento en la expresión del receptor de AMPA observado con la cocaína no ocurre en ratas sensibilizadas con anfetamina, lo que lleva a la hipótesis de diferentes efectos funcionales de las proyecciones de glutamato al núcleo accumbens durante la abstinencia de la cocaína frente a la anfetamina (Nelson et al, 2009).
De acuerdo con los resultados de la alteración de la neurotransmisión de glutamato en ratas sensibilizadas con cocaína, los estudios de microdiálisis y microinyección han demostrado que después de la cocaína crónica se produce una disminución de la liberación basal de glutamato, pero se sensibiliza la liberación de glutamato sináptico durante el restablecimiento de la búsqueda de drogas extinguida en ratas (Kalivas y O ‘ Brien, 2008; McFarland et al, 2003). Se ha hipotetizado que esta desregulación del glutamato es causada por la disminución de la función del intercambiador de cistina-glutamato (Baker et al, 2003) y la desensibilización del receptor de glutamato mGlu2 / 3 metabotrópico. Los niveles basales más bajos de glutamato, combinados con una mayor liberación de glutamato sináptico de la activación de aferentes de la corteza prefrontal al núcleo accumbens, se hipotetizan como resultado de un impulso para participar en la búsqueda de drogas (Kalivas, 2004).
Estos efectos sinápticos de larga duración producen tanto una disminución en la neurotransmisión de glutamato durante la administración crónica del fármaco como un aumento persistente en la eficacia de la neurotransmisión sináptica glutamatérgica durante el restablecimiento después de la retirada. Estos cambios dinámicos pueden promover la excitación celular, que se ha hipotetizado como un sustrato importante para la sensibilización y el aprendizaje relacionado con las drogas en el estado adictivo (Kauer y Malenka, 2007; Wolf et al, 2004).
Como lo sugirieron anteriormente los modelos animales, la magnitud de la liberación de dopamina estriatal (particularmente en su aspecto ventral) en los humanos se correlaciona positivamente con la respuesta hedónica a la mayoría de las drogas de abuso, incluida la anfetamina (Drevets y otros, 2001), la cocaína (Volkow y otros, 1997a), metilfenidato (Volkow et al, 2002) y nicotina (Sharma y Brody, 2009). Es probable que los aumentos dependientes de las drogas, rápidos y suprafisiológicos en la dopamina imiten los cambios de dopamina inducidos por la activación de células de dopamina fásica que se produce en respuesta a estímulos salientes, categorizando así la experiencia de la droga como una que es altamente destacada, un resultado experiencial que llama la atención y promueve la excitación, el aprendizaje condicionado y la motivación (Volkow et al, 2004b). Sobre la base de los hallazgos en animales de laboratorio, se postula que la exposición frecuente a estas respuestas farmacológicas en los drogadictos da como resultado la recalibración de los umbrales de activación (recompensa) de dopamina para refuerzos naturales.
Por lo tanto, se puede imaginar el desarrollo de un cambio en la activación de las neuronas de dopamina mesolímbicas que comienza con una administración del fármaco, se desarrolla primero en LTP en el VTA, luego en el núcleo accumbens, y posteriormente, mediante bucles de realimentación, se engancha al estriado dorsal. Además, pueden seguir los cambios a largo plazo en la CeA y la corteza prefrontal medial, y combinados con la desregulación de los sistemas de estrés cerebral (ver a continuación) pueden proporcionar un poderoso impulso para el comportamiento de búsqueda de drogas incluso meses después de la retirada de la droga (Figura 4 y 5).
Estriado ventral / Estriado dorsal / tálamo: voluntario a la búsqueda habitual de drogas
La hipótesis de que el circuito estriado dorsal tiene un papel clave en el desarrollo del uso compulsivo habitual de cocaína se apoya en datos que muestran la importancia para el estriado dorsal en el aprendizaje de hábitos de estímulo-respuesta (Yin et al, 2005) y estudios de microdiálisis que demuestran que la cocaína prolongada buscando una mayor liberación de dopamina en el estriado dorsal pero no en el estriado ventral (Ito et al, 2002). La desconexión del estriado ventral del estriado dorsal en ratas autoadministradas con cocaína en un programa de segundo orden solo mostró un déficit en animales con una ingesta «compulsiva» bien establecida pero no en animales que adquirieron recientemente el programa de segundo orden (Belin y Everitt, 2008). Por lo tanto, la hipótesis es que la adicción a las drogas representa cambios en las estructuras asociativas para convertirse en automáticas o habituales e involucra un compromiso gradual de los mecanismos estriatales dorsales.
Los estudios en animales han sugerido enérgicamente que, con la exposición repetida al fármaco, los estímulos neutros asociados con el fármaco pueden adquirir la capacidad de aumentar la dopamina por sí mismos. Los estudios de imágenes cerebrales confirmaron esto en humanos adictos (Volkow et al, 2008a; Heinz et al, 2004). Estos estudios demostraron que las señales asociadas con el fármaco indujeron aumentos de dopamina en el cuerpo estriado dorsal (caudado y putamen), un efecto que se correlacionaba con los autoinformes de ansia. El hecho de que la magnitud de los aumentos de dopamina desencadenados por las señales se asoció con el grado de severidad de la adicción destaca la importancia de estas respuestas condicionadas de dopamina en el proceso de adicción a las drogas en los seres humanos.
Los estudios clínicos también han demostrado que los aumentos de dopamina lentos en el cuerpo estriado inducidos por la administración aguda de metilfenidato oral no provocan ansia en los consumidores de cocaína a menos que estén acoplados a señales asociadas a las drogas (Volkow et al, 2008a). Lo más probable es que esto refleje el hecho de que el ansia se debe a los rápidos aumentos de dopamina logrados con la cocción fásica de dopamina, en oposición a los aumentos lentos de dopamina logrados con la cocción de dopamina tónica y en el experimento con metilfenidato oral. De hecho, la administración intravenosa de metilfenidato, que resulta en un rápido aumento de la dopamina, induce un deseo intenso.
Los estudios de imágenes cerebrales también han demostrado que, en sujetos adictos a las drogas, estos procesos involucran la corteza orbitofrontal, una región del cerebro implicada en la atribución y motivación de la saliencia, cuya interrupción produce compulsividad, y es una región del cerebro con grandes proyecciones al estriado dorsal. El giro cingulado también está involucrado y es una región del cerebro implicada en el control inhibitorio y la resolución de conflictos, cuya interrupción produce impulsividad (Volkow et al, 2004b). Además, en los sujetos adictos a la cocaína, pero no sin adicción, la administración intravenosa de metilfenidato, que según los consumidores de cocaína tienen efectos similares a los de la cocaína, activó las cortezas prefrontales orbitales y mediales, y esta activación se asoció con el deseo de cocaína (Volkow et al. al, 2005). Del mismo modo, en los sujetos con la marihuana adicto, pero no en los individuos no adictos, la administración aguda de Δ 9 -THC activa la corteza obitofrontal (Volkow et al, 1996a). La activación de la corteza obitofrontal y el giro cingulado también se desencadena mediante señales condicionadas que predicen la recompensa y desencadenan el deseo (McClernon et al, 2009). Curiosamente, estas son regiones que regulan la activación y liberación de células de dopamina, que se han postulado como necesarias para aumentar los valores motivacionales de incentivo de las drogas en individuos adictos (reflejando una hipótesis basada en estudios en animales; Volkow et al, 1999). Cuando se combinan, estas observaciones sugieren fuertemente que los aumentos de dopamina asociados con señales condicionadas no son respuestas primarias, sino más bien el resultado de la estimulación por retroalimentación de las células de dopamina, muy probablemente aferentes glutamatérgicos de la corteza prefrontal y / o amígdala. Sobre la base de estos hallazgos, se ha planteado la hipótesis de que la activación de la corteza obitofrontal, con aumentos concomitantes de la dopamina producida por la droga, contribuye al consumo compulsivo de drogas que caracteriza a los atracones de drogas en individuos adictos (Volkow et al, 2007).
De hecho, los estudios de neuroimagen humana muestran que la corteza prefrontal (orbitofrontal, prefrontal medial, prelímbica / cingulada) y la amígdala basolateral son fundamentales para el anhelo en los seres humanos inducido por drogas y señales (Franklin et al, 2007). En las regiones prefrontales (por ejemplo, giro cingulado y la corteza obitofrontal), estos cambios se han asociado con una reducción de la dopamina D estriatal 2 disponibilidad del receptor observada en sujetos adictos (Heinz et al, 2004; Volkow et al, 1993, 2001a, 2007). Estas asociaciones podrían reflejar una interrupción de las regiones cerebrales frontales secundaria a cambios en la actividad de la dopamina del estriado, o alternativamente podrían reflejar una interrupción primaria de las regiones frontales que regulan la actividad de las células de la dopamina. De hecho, un estudio PET reciente proporcionó pruebas de que las regiones cerebrales prefrontales regulan el valor de las recompensas al modular los aumentos de dopamina en el estriado ventral, un mecanismo regulador que se vuelve disfuncional en los individuos adictos (Volkow et al, 2007).
Por lo tanto, la neurotransmisión concomitante de dopamina y glutamato en el cuerpo estriado dorsal, una región implicada en el aprendizaje del hábito y la iniciación de la acción, está involucrada en el deseo de depender del contexto. Como tal, el cuerpo estriado dorsal puede ser un componente fundamental de la adicción (Volkow et al, 2006). La investigación sobre estrategias novedosas para inhibir las respuestas de dopamina y glutamato condicionadas por señales es un enfoque importante de los esfuerzos actuales de desarrollo de medicamentos.
El tálamo no se ha estudiado tan extensamente en el contexto de la adicción. Sin embargo, debido a su función integradora en la regulación de la excitación y la modulación de la atención, esta región se ha visto cada vez más implicada en el proceso de adicción. Por ejemplo, la administración intravenosa de una droga estimulante en los consumidores de cocaína, pero no en los controles, incrementó la neurotransmisión de dopamina en el tálamo, un efecto asociado con el deseo (Volkow et al, 1997a). En contraste, en comparación con los controles, los consumidores de cocaína muestran una hipoactivación del tálamo, posiblemente reflejando déficits noradrenérgicos y / o dopaminérgicos, cuando realizan una tarea cognitiva (Tomasi et al, 2007b). De manera similar, se informó que el tálamo mostraba una activación atenuada mientras realizaba una tarea cognitiva visual en fumadores expuestos a la nicotina (Sharma y Brody, 2009). Estos resultados sugieren que las anomalías talámicas en los consumidores de cocaína pueden contribuir no solo a las deficiencias en el procesamiento sensorial y la atención, sino también al deseo. Curiosamente, los cambios en la transmisión de dopamina en el tálamo y el estriado parecen estar involucrados en el deterioro del rendimiento cognitivo (por ejemplo, atención visual y memoria de trabajo) que inexorablemente sigue a un período de privación del sueño (Volkow et al, 2008b). Por lo tanto, se justifica una mayor investigación que se basa en los datos preliminares disponibles.
Corteza frontal dorsolateral, corteza frontal inferior, hipocampo: control cognitivo, gratificación retardada y memoria
La adicción también implica perturbaciones en los procesos cognitivos y emocionales regulados corticalmente, que causan la sobrevaluación de los reforzadores de medicamentos a expensas de la subvaloración de los reforzadores naturales y los déficits en el control inhibitorio de las respuestas a los medicamentos (Goldstein y Volkow, 2002). Como resultado, se cree que un sistema prefrontal de bajo rendimiento es crucial para el proceso de adicción.
Uno de los componentes de dicho sistema es el control de impulsos, que se encuentra entre los factores de riesgo cognitivo más sólidos para los trastornos por uso de sustancias. La cocaína parece tener un efecto directo en la neurobiología subyacente al control de impulsos. Después de una inyección intravenosa de cocaína, los usuarios de cocaína en realidad mostraron una mejoría en la tarea de inhibición de la respuesta motora y una mayor activación concomitante en sus cortes dorsolateral derecho e inferior frontal (Garavan et al, 2008). Debido a que estas áreas se consideran importantes en el control de impulsos, esta observación sugiere que algunos de los efectos agudos de la cocaína podrían, de hecho, mediar una reversión transitoria de la hipofunción crónica en los circuitos de control de impulsos.
Otra función importante que reside en las áreas frontocorticales es la capacidad de elegir entre recompensas pequeñas e inmediatas en comparación con las recompensas grandes pero diferidas, que se pueden medir utilizando una tarea de descuento diferido. Un estudio reciente descubrió que los volúmenes de materia gris dorsolateral y córtex frontal inferolateral se correlacionaban inversamente con la preferencia por la gratificación inmediata durante la toma de decisiones (Bjork et al, 2009). Este hallazgo sugiere que las anomalías en las regiones frontocorticales pueden subyacer a la incapacidad de retrasar la gratificación, un rasgo que es característico de la adicción y otros trastornos psiquiátricos.
Los sustratos neurales de la memoria y el aprendizaje condicionado se encuentran entre los circuitos principales que experimentan neuroadaptaciones anormales en respuesta a la exposición crónica a drogas (Volkow et al, 2004a). Se ha propuesto que diferentes sistemas de memoria se involucren en la adicción a las drogas, incluido el aprendizaje de incentivo condicionado (a través del núcleo accumbens y la amígdala), el aprendizaje del hábito (a través del caudado y el putamen) y la memoria declarativa (a través del hipocampo; White, 1996) que es el tema central de esta sección.
Durante la última década, muchos estudios provocativos con animales han sugerido que las drogas adictivas pueden alterar la neurogénesis en el hipocampo adulto (Canales, 2007). Se demostró que el daño al subículo ventral del hipocampo afecta la autoadministración de cocaína en ratas (Caine et al, 2001). Dichas observaciones han proporcionado información sobre la posible participación de un hipocampo desregulado en la adicción humana. Esta hipótesis es una extensión del conocimiento actual porque el hipocampo se considera en general como importante en el condicionamiento contextual, es decir, en el procesamiento de señales contextuales mediante las cuales se puede acceder y recuperar las memorias. De hecho, desde hace tiempo se reconoce que la memoria declarativa está involucrada en el aprendizaje y la vinculación de las condiciones o circunstancias afectivas con las experiencias de consumo de drogas. Estudios con TEP y resonancia magnética funcional han demostrado que el ansia provocada por el estímulo, así como la intoxicación aguda, activan el hipocampo y la amígdala (Volkow et al, 2004a). Por ejemplo, el deseo que experimentan los consumidores de cocaína cuando están expuestos a estímulos relacionados con las drogas está acompañado por aumentos en el flujo sanguíneo en una región distribuida implicada en varias formas de memoria, incluida la amígdala (Childress y otros, 1999; Grant y otros, 1996; Kilts et al, 2001) e hipocampo (Kilts et al, 2001).
Por lo tanto, los nuevos enfoques para interrumpir la reconsolidación de la memoria pueden ayudar a erosionar las fuertes asociaciones entre el contexto y la droga (Lee, 2008; Lee et al, 2005). Curiosamente, los bloqueadores β ya han demostrado una capacidad prometedora para inhibir las respuestas condicionadas tanto a los reforzadores naturales como a los estímulos aversivos (Miranda et al, 2003). Además, los resultados de un estudio más reciente sugieren que las respuestas condicionadas inducidas por drogas también pueden ser sensibles al tratamiento con β bloqueante (Milton et al, 2008). Del mismo modo, también parece justificada una mayor investigación sobre las drogas que mejoran el GABA. La estimulación GABAérgica, que puede atenuar el condicionamiento pavloviano, parece interrumpir la respuesta a las drogas de abuso en animales (Volkow et al, 2004a) y puede ser una estrategia útil para tratar la adicción en humanos (Dewey et al, 1998).
Amígdala extendida: vías de refuerzo negativo
El uso compulsivo de drogas definido por una mayor ingesta de drogas con acceso extendido está acompañado por una perturbación crónica en la homeostasis de recompensa cerebral utilizando medidas de umbrales de recompensa de estimulación cerebral. La exposición diferencial a la autoadministración de medicamentos tiene efectos dramáticos en los umbrales de recompensa que aumentan progresivamente (es decir, disminuyen la recompensa) en ratas de acceso extendido, pero no de acceso limitado, en sesiones sucesivas de autoadministración (Ahmed et al, 2002; Kenny et al, 2006; Wee et al, resultados no publicados). Los animales con acceso extendido a la cocaína son más sensibles al bloqueo de la autoadministración por los antagonistas de la dopamina y los agonistas parciales (Ahmed y Koob, 2004; Wee et al, 2007), y la dosis parcial de buprenorfina del agonista opioide redujo de forma dependiente la autoadministración de heroína en ratas dependientes de opioides de acceso extendido (Chen et al, 2006b), lo que sugiere que la reversión de los déficits de recompensa puede mitigar los impulsos motivacionales de la adicción a las drogas. Este mecanismo podría ser la base del beneficio del tratamiento con metadona y buprenorfina en la adicción a la heroína.
Como se señaló anteriormente, los antagonistas de CRF bloquearon los efectos ansiogénicos y aversivos de la abstinencia de drogas, y la retirada de todas las drogas de abuso activó la CRF en la CeA. Estas observaciones llevaron a la hipótesis de que la activación de CRF, específicamente CRF extrahipotalámico en el CeA, contribuyó a la impulsividad del estado motivacional desde la perspectiva del refuerzo negativo (Koob y Le Moal, 2008). Por lo tanto, uno podría predecir que el bloqueo de los sistemas de estrés cerebral en modelos animales de acceso extendido a las drogas puede bloquear la motivación de una ingesta excesiva de drogas. Los antagonistas de CRF bloquearon selectivamente el aumento de la autoadministración de drogas asociadas con el acceso extendido a la autoadministración intravenosa de cocaína, nicotina (Koob, 2008), heroína (Greenwell et al, 2009), y el alcohol (Koob, 2008). Un ejemplo particularmente dramático de los efectos motivacionales de CRF en la amígdala extendida en dependencia puede observarse en modelos animales de autoadministración de etanol en animales dependientes en los que un antagonista del péptido CRF 1/2 inyectado en la amígdala bloqueó el aumento de la autoadministración de etanol durante la abstinencia. (Funk et al, 2006; Koob, 2008).
Aunque menos desarrollada, la evidencia sugiere la participación de los sistemas de norepinefrina en la amígdala extendida en el estado motivacional negativo y una mayor autoadministración asociada con la dependencia (Koob, 2009b). Consistente con el papel del sistema opioide dynorphin- κ en los efectos aversivos de la retirada del fármaco, un antagonista κ -opioide bloqueó el consumo excesivo de alcohol asociado con la extracción de etanol en ratas dependientes y bloqueó selectivamente el rendimiento de la relación progresiva aumentada en ratas con acceso extendido cocaína (Koob, 2009b; Wee et al, 2009).
El neuropéptido Y tiene propiedades dramáticas de tipo ansiolítico localizadas en la amígdala y se ha planteado la hipótesis de que tiene efectos opuestos al CRF en el estado motivacional negativo de abstinencia de drogas de abuso (Heilig y otros, 1994; Heilig y Koob, 2007). La administración de NPY intracerebroventricular bloqueó el aumento de la ingesta de drogas asociada con la dependencia del etanol (Thorsell et al, 2005a, 2005b). La inyección de NPY en la CeA (Gilpin et al, 2008) y la expresión potenciada por vector viral de NPY en la CeA también bloquearon el aumento de la ingesta de medicamentos asociada con la dependencia del etanol (Thorsell et al, 2007).
Por lo tanto, los aumentos de CRF en el CeA que se producen con la abstinencia aguda de los medicamentos tienen una importancia motivacional no solo para los efectos de ansiedad / aversivos de la abstinencia aguda, sino también para la mayor ingesta de medicamentos asociada con la dependencia. La abstinencia aguda también puede aumentar la liberación de norepinefrina en el BNST y la dinorfina en el núcleo accumbens, lo que puede contribuir al estado emocional negativo asociado con la dependencia. La disminución de la actividad del NPY en el CeA también puede contribuir al estado de ansiedad asociado con la dependencia del etanol. La activación de los sistemas de estrés cerebral (CRF, norepinefrina, dinorfina), combinada con la inactivación de los sistemas antiestrés cerebrales (NPY) en la amígdala extendida puede provocar una fuerte desregulación emocional con un significado motivacional para la adicción. (Koob, 2008). Dicha desregulación puede ser una contribución significativa a los procesos del oponente entre sistemas que ayudan a mantener la dependencia y también prepara el escenario para cambios de estado más prolongados en la emocionalidad, como la abstinencia prolongada.
La investigación sobre los mecanismos de refuerzo negativo en la adicción humana ha sido muy limitada. Con la cocaína, por ejemplo, se demostró que la amígdala y la corteza orbitofrontal lateral estaban activadas por infusiones inesperadas, pero no esperadas de cocaína en consumidores de cocaína activos (Kufahl et al, 2008), pero la abstinencia de la cocaína se asoció con grandes reducciones en la actividad de la proyección de dopamina. regiones, incluida la amígdala (Tomasi et al, 2007a). En aparente contraste, la abstinencia de fumar se asoció con un aumento del flujo sanguíneo cerebral en la amígdala extendida, entre otras regiones (Wang et al, 2007), mientras que un aerosol nasal de nicotina redujo el flujo sanguíneo cerebral regional en la amígdala derecha y la corteza temporal anterior izquierda de los fumadores habituales sometidos a 12 h de privación de fumar (Zubieta et al, 2001).
La amígdala puede ser igualmente importante para procesar la recompensa positiva (Murray, 2007) y la expectativa de recompensa (Holland y Gallagher, 2004), similar al procesamiento de la recompensa negativa. Particularmente interesante en el contexto de la investigación de imágenes cerebrales será comprender la función de la amígdala para generar la ansiedad y las emociones negativas que se ven con frecuencia durante la abstinencia. Un informe reciente destacó la importancia de la adicción al circuito interoceptivo que más probablemente interactúa con la amígdala extendida y el estriado ventral. El estudio mostró que los fumadores con daño en la ínsula (pero no los fumadores con lesiones extrainsulares) podían dejar de fumar fácilmente y sin experimentar antojos ni recaídas (Naqvi et al, 2007). La ínsula, particularmente sus regiones más anteriores, está conectada recíprocamente a varias regiones límbicas (p. Ej., La corteza prefrontal ventromedial, la amígdala y el estriado ventral) y parece tener una función interoceptiva, integrando la información autónoma y visceral con emoción y motivación y proporcionando conciencia de estos impulsos (Naqvi y Bechara, 2009). De hecho, los estudios de lesiones cerebrales sugieren que la corteza y la ínfrecha ventromedial son componentes necesarios de los circuitos distribuidos que apoyan la toma de decisiones emocionales (Clark et al, 2008). De acuerdo con esta hipótesis, muchos estudios de imagen muestran una activación diferencial en la ínsula durante el deseo (Naqvi y Bechara, 2009). Se ha sugerido que la reactividad de esta región del cerebro sirve como biomarcador para ayudar a predecir la recaída.
Objetivos moleculares para la neuroplasticidad: atracón / intoxicación, retiro/ efecto negativo, y preocupación/ anticipación (craving)
El enfoque de la presente revisión está en los neurocircuitos de la adicción. Sin embargo, paralelamente a la neuroplasticidad de los neurocircuitos están los cambios moleculares que ocurren en estas mismas estructuras. La exposición crónica a opiáceos y cocaína conduce a la activación de la proteína de unión al elemento de respuesta al monofosfato de adenosina cíclica (CREB) en el núcleo accumbens y CeA (Shaw-Lutchman et al, 2002; Edwards et al, 2007). CREB puede ser fosforilada por la proteína quinasa A y por la proteína quinasa regulada por factores de crecimiento, lo que lo sitúa en un punto de convergencia para varias vías de mensajería intracelular que pueden regular la expresión génica. La activación de CREB en el núcleo accumbens con fármacos psicoestimulantes está relacionada con los síntomas motivadores de la abstinencia psicoestimulante, como disforia, posiblemente a través de la inducción del péptido opioide dinorfina, que se une a los receptores opioides κ y se ha hipotetizado para representar un mecanismo de motivación Tolerancia y dependencia (Nestler, 2005). La activación repetida de CREB promueve la expresión de la dinorfina en el núcleo accumbens, que a su vez disminuye la actividad dopaminérgica, lo que puede contribuir a estados emocionales negativos. La quinasa regulada por señal extracelular es otro elemento clave de la señalización intracelular que se considera un componente clave en la plasticidad asociada con la administración repetida de cocaína, específicamente la sensibilización del comportamiento, la recompensa de la cocaína y los aumentos dependientes del tiempo en la búsqueda de cocaína después del retiro (es decir, el efecto de incubación; Lu et al, 2006; Li et al, 2008).
Otro objetivo molecular para regular la plasticidad que conduce a la adicción es la desregulación del intercambio de cistina-glutamato, que se cree que promueve la señalización patológica de glutamato relacionada con varios componentes del ciclo de adicción. Aquí, la administración repetida de cocaína atenúa el intercambio de cistina-glutamato, lo que lleva a una reducción del glutamato inducido por cocaína basal y aumentado en el núcleo accumbens que persiste durante al menos 3 semanas después del último consumo de cocaína (Baker et al, 2003). Lo más convincente es la observación de que el tratamiento con N-acetilcisteína, al activar el intercambio de cistina-glutamato, evitó la intensificación inducida por la cocaína y la sensibilización del comportamiento, restableció la capacidad de inducir LTP y depresión a largo plazo en el núcleo accumbens y limitó el restablecimiento en animales y la reactividad condicionada a las señales de las drogas en humanos (Moussawi et al, 2009; LaRowe et al, 2007; Madayag et al, 2007).
CREB y otros mensajeros intracelulares pueden activar los factores de transcripción, que pueden cambiar la expresión génica y producir cambios a largo plazo en la expresión de proteínas y, como resultado, la función neuronal. Aunque la administración aguda de drogas de abuso puede causar una activación rápida (dentro de unas horas) de miembros de la familia de proteínas Fos, como c-fos, FosB, Fra-1 y Fra-2 en el núcleo accumbens, otros factores de transcripción, isoformas de ΔFosB, una forma altamente estable de FosB, se ha demostrado que se acumula durante largos períodos de tiempo (días) con la administración repetida de medicamentos (Nestler, 2005). Los animales con ΔFosB activado tienen una sensibilidad exagerada a los efectos gratificantes de las drogas de abuso, y ΔFosB puede ser un «cambio» molecular sostenido que ayuda a iniciar y mantener un estado de adicción (McClung et al, 2004). Queda por determinar si (y cómo) dichos factores de transcripción influyen en la función de los sistemas de estrés cerebral, como el CRF y los descritos anteriormente.
Resumen y conclusiones
En resumen, las múltiples regiones y circuitos cerebrales están alterados en la adicción a las drogas y es probable que contribuyan de manera diferente al complejo fenotipo observado en individuos adictos (Figura 5). Aunque algunas de estas anomalías funcionales pueden estar presentes en mayor o menor medida en todas las clases de adicciones a las drogas, algunos de los cambios pueden ser específicos de ciertos tipos de drogas. Por ejemplo, los decrementos de larga duración en el DAT en el estriado se observan en la metanfetamina, pero no en las adicciones al alcohol o la cocaína. A la inversa, se observan disminuciones en los receptores de dopamina D2 en el cuerpo estriado en sujetos adictos a todas las drogas de abuso que se han investigado, y se ha observado una mayor activación de los sistemas de estrés cerebral como el CRF en modelos animales durante el retiro agudo para todos los tipos de las drogas. Es importante destacar que las anormalidades neuronales que se manifiestan en un individuo adicto y que pueden descubrirse mediante estudios de imagen y / o neuropsicofarmacológicos son un reflejo de no solo una trayectoria crónica de exposición a la sustancia, sino también de las variaciones específicas de un individuo en las características de genética, desarrollo y entorno.
Líneas futuras de investigación
Los avances descritos anteriormente señalan el camino hacia direcciones futuras para la investigación en neurocircuitos de la adicción en el mismo marco conceptual de borrachera / intoxicación, abstinencia / afecto negativo y preocupación / anticipación. Los ricos recursos de las neurociencias modernas aplicadas a la neurobiología de la adicción ofrecen una oportunidad no solo para comprender los neurocircuitos del proceso de adicción, sino también para proporcionar las claves para comprender la vulnerabilidad y brindar tratamiento para esta enfermedad devastadora.
En la etapa de embriaguez / intoxicación del ciclo de adicción, la neuroplasticidad que comienza con un cambio en la activación de las neuronas de dopamina mesolímbicas durante la exposición inicial al fármaco se traduce en compromiso del estriado dorsal, interrupción de la función del sistema frontal y reclutamiento de sistemas de estrés cerebral y los resultados en un poderoso impulso residual para el comportamiento de búsqueda de drogas, incluso meses después de la retirada, queda por determinar. Por ejemplo, ¿cuál es la relación entre la vulnerabilidad a la impulsividad y la compulsividad posterior en la neuroplasticidad de los circuitos descritos anteriormente? Dichos estudios futuros pueden involucrar enfoques genéticos moleculares que van desde la reproducción selectiva hasta la regulación positiva o la eliminación de mecanismos moleculares dentro de circuitos cerebrales específicos utilizando tecnología de ARN de horquilla corta.
En la etapa de retiro / efecto negativo, el compromiso de los sistemas de estrés cerebral, como el CRF, en modelos animales debe extenderse a otros sistemas interactivos de estrés cerebral y explorarse en estudios en humanos. Actualmente se están explorando numerosos otros sistemas de neurotransmisores que interactúan con el sistema de estrés cerebral, como la dinorfina, el NPY, la sustancia P, la nociceptina y la orexina. Prácticamente inexplorados en esta etapa están los estudios de imágenes en humanos de este componente del ciclo de adicción y las imágenes en humanos de los sistemas de neurotransmisores cerebrales implicados en los aspectos motivacionales de la retirada de fármacos. El desarrollo de nuevos ligandos radioactivos para estudios de imágenes en humanos que se unen a los receptores de los sistemas neurotransmisores anteriores sería un gran impulso para el campo.
En la etapa de preocupación / anticipación, los estudios de neuroimagen humana muestran que la corteza prefrontal (orbitofrontal, prefrontal medial, prelímbica / cingulada) y la amígdala basolateral son fundamentales en el deseo de drogas e indicios. Queda por determinar si dichas asociaciones reflejan una interrupción de las regiones cerebrales frontales secundarias a cambios en la actividad de la dopamina del estriado, o alternativamente reflejan una interrupción primaria de las regiones frontales que regulan la actividad de las células de la dopamina. Los nuevos enfoques para el estudio de la reconsolidación de la memoria pueden ayudar a dilucidar las fuertes asociaciones entre el contexto y el fármaco. La importancia en la adicción del circuito interoceptivo que involucra la ínsula y otras regiones que probablemente se interconectan con la amígdala extendida y el estriado ventral aún no se ha determinado. La reactividad de estos circuitos cerebrales puede servir como biomarcador para ayudar a predecir la recaída y ayudar a predecir la eficacia del tratamiento. Los estudios post mortem en humanos, los estudios de laboratorio en humanos y los estudios de neurocircuitos en modelos animales paralelos probablemente darán resultados prometedores en este dominio. Finalmente, solo se están dilucidando los cambios moleculares y genéticos que transmiten los cambios en la actividad de los neurocircuitos en las tres etapas del ciclo de adicción descrito anteriormente. Los cambios en los sistemas reguladores del transmisor, los factores de transcripción e incluso la regulación de genes a nivel epigenético pueden explicar cómo se desregulan los circuitos, permanecen desregulados y proporcionan vulnerabilidad a la desregulación inicialmente o durante mucho tiempo en la abstinencia. En última instancia, los objetivos neurobiológicos dilucidados a través del marco de los neurocircuitos de la adicción proporcionarán objetivos para identificar la vulnerabilidad genética en la población humana, y la vulnerabilidad genética en los estudios humanos puede identificar nuevos objetivos para ser explorados a nivel mecanicista en estudios con animales.
Referencias
Aharonovich E, Hasin DS, Brooks AC, Liu X, Bisaga A, Nunes EV (2006). Cognitive deficits predict low treatment retention in cocaine dependent patients. Drug Alcohol Depend 81: 313–322.
Ahmed SH, Kenny PJ, Koob GF, Markou A (2002). Neurobiological evidence for hedonic allostasis associated with escalating cocaine use. Nat Neurosci 5: 625–626.
Ahmed SH, Koob GF (1998). Transition from moderate to excessive drug intake: change in hedonic set point. Science 282: 298–300. This study showed that rats given extended access to cocaine escalate intake and show behavior consistent with an increase in hedonic set point (lower reward) for the drug.
Ahmed SH, Koob GF (2004). Changes in response to a dopamine antagonist in rats with escalating cocaine intake. Psychopharmacology 172: 450–454.
Ahmed SH, Walker JR, Koob GF (2000). Persistent increase in the motivation to take heroin in rats with a history of drug escalation. Neuropsychopharmacology 22: 413–421.
Alheid GF, De Olmos JS, Beltramino CA (1995). Amygdala and extended amygdala. In: Paxinos G (ed). The Rat Nervous System. Academic Press: San Diego. Pp 495–578.
Allen TJ, Moeller FG, Rhoades HM, Cherek DR (1998). Impulsivity and history of drug dependence. Drug Alcohol Depend 50: 137–145.
American Psychiatric Association (1994). Diagnostic and Statistical Manual of Mental Disorders 4th edn. American Psychiatric Press: Washington, DC.
American Psychiatric Association (2000). Diagnostic and Statistical Manual of Mental Disorders 4th edn, text revision American Psychiatric Press: Washington, DC.
Arroyo M, Markou A, Robbins TW, Everitt BJ (1998). Acquisition, maintenance and reinstatement of intravenous cocaine self-administration under a second-order schedule of reinforcement in rats: effects of conditioned cues and continuous access to cocaine. Psychopharmacology 140: 331–344.
Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S et al (2003). Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 6: 743–749.
Baker TB, Morse E, Sherman JE (1987). The motivation to use drugs: a psychobiological analysis of urges. In: River PC (ed). Alcohol and Addictive Behavior (series title: Nebraska Symposium on Motivation, vol 34). University of Nebraska Press: Lincoln, NE. pp 257–323.
Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton KT (1991). CRF antagonist reverses the ‘anxiogenic’ response to ethanol withdrawal in the rat. Psychopharmacology 103: 227–232.
Barr AM, Phillips AG (1999). Withdrawal following repeated exposure to d-amphetamine decreases responding for a sucrose solution as measured by a progressive ratio schedule of reinforcement. Psychopharmacology 141: 99–106.
Belin D, Everitt BJ (2008). Cocaine seeking habits depend upon dopaminedependent serial connectivity linking the ventral with the dorsal striatum. Neuron 57: 432–441. This study showed that the interactions between the ventral and dorsal striatum are critical for the development of compulsive-like cocaineseeking behavior.
Ben-Shahar O, Ahmed SH, Koob GF, Ettenberg A (2004). The transition from controlled to compulsive drug use is associated with a loss of sensitization. Brain Res 995: 46–54.
Bolla KI, Eldreth DA, London ED, Kiehl KA, Mouratidis M, Contoreggi C et al (2003). Orbitofrontal cortex dysfunction in abstinent cocaine abusers performing a decision-making task. Neuroimage 19: 1085–1094.
Bonci A, Bernardi G, Grillner P, Mercuri NB (2003). The dopamine-containing neuron: maestro or simple musician in the orchestra of addiction? Trends Pharmacol Sci 24: 172–177.
Boudreau AC, Wolf ME (2005). Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci 25: 9144–9151.
Briand LA, Flagel SB, Garcia-Fuster MJ, Watson SJ, Akil H, Sarter M et al (2008a). Persistent alterations in cognitive function and prefrontal dopamine D2 receptors following extended, but not limited, access to self-administered cocaine. Neuropsychopharmacology 33: 2969–2980.
Briand LA, Gross JP, Robinson TE (2008b). Impaired object recognition following prolonged withdrawal from extended-access cocaine self-administration. Neuroscience 155: 1–6.
Caine SB, Heinrichs SC, Coffin VL, Koob GF (1995). Effects of the dopamine D-1 antagonist SCH 23390 microinjected into the accumbens, amygdala or striatum on cocaine self-administration in the rat. Brain Res 692: 47–56.
Caine SB, Humby T, Robbins TW, Everitt BJ (2001). Behavioral effects of psychomotor stimulants in rats with dorsal or ventral subiculum lesions: locomotion, cocaine self-administration, and prepulse inhibition of startle. Behav Neurosci 115: 880–894.
Caine SB, Thomsen M, Gabriel KI, Berkowitz JS, Gold LH, Koob GF et al (2007). Lack of self-administration of cocaine in dopamine D1 receptor knock-out mice. J Neurosci 27: 13140–13150.
Calu DJ, Stalnaker TA, Franz TM, Singh T, Shaham Y, Schoenbaum G (2007). Withdrawal from cocaine self-administration produces long-lasting deficits in orbitofrontal-dependent reversal learning in rats. Learn Mem 14: 325–328.
Canales JJ (2007). Adult neurogenesis and the memories of drug addiction. Eur Arch Psychiatry Clin Neurosci 257: 261–270.
Chait LD (1994). Reinforcing and subjective effects of methylphenidate in humans. Behav Pharmacol 5: 281–288.
Chao SZ, Ariano MA, Peterson DA, Wolf ME (2002). D1 dopamine receptor stimulation increases GluR1 surface expression in nucleus accumbens neurons. J Neurochem 83: 704–712.
Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM et al (2008). Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron 59: 288–297.
Chen R, Tilley MR, Wei H, Zhou F, Zhou FM, Ching S et al (2006a). Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc Natl Acad Sci USA 103: 9333–9338.
Chen SA, O’Dell L, Hoefer M, Greenwell TN, Zorrilla EP, Koob GF (2006b). Unlimited access to heroin self-administration: independent motivational markers of opiate dependence. Neuropsychopharmacology 31: 2692–2707 (corrigendum: 31: 2802).
Childress AR, McLellan AT, Ehrman R, O’Brien CP (1988). Classically conditioned responses in opioid and cocaine dependence: a role in relapse?. In: Ray BA (ed). Learning Factors in Substance Abuse (series title: NIDA Research Monograph, vol 84). National Institute on Drug Abuse: Rockville, MD. pp 25–43.
Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP (1999). Limbic activation during cue-induced cocaine craving. Am J Psychiatry 156: 11–18.
Clark L, Bechara A, Damasio H, Aitken MR, Sahakian BJ, Robbins TW (2008). Differential effects of insular and ventromedial prefrontal cortex lesions on risky decision-making. Brain 131: 1311–1322.
Collins RJ, Weeks JR, Cooper MM, Good PI, Russell RR (1984). Prediction of abuse liability of drugs using IV self-administration by rats. Psychopharmacology 82: 6–13.
Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y et al (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454: 118–121.
Creese I, Iversen SD (1974). The role of forebrain dopamine systems in amphetamine-induced stereotyped behavior in the rat. Psychopharmacology 39: 345–357.
Crow TJ (1973). Catecholamine-containing neurones and electrical self-stimulation: 2. A theoretical interpretation and some psychiatric implications. Psychol Med 3: 66–73.
de Witte P, Littleton J, Parot P, Koob G (2005). Neuroprotective and abstinencepromoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs 19: 517–537.
Delfs JM, Zhu Y, Druhan JP, Aston-Jones G (2000). Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature 403: 430–434.
Deroche-Gamonet V, Belin D, Piazza PV (2004). Evidence for addiction-like behavior in the rat. Science 305: 1014–1017.
Dewey SL, Morgan AE, Ashby Jr CR, Horan B, Kushner SA, Logan J et al (1998). A novel strategy for the treatment of cocaine addiction. Synapse 30: 119–129.
Di Chiara G, Imperato A (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 85: 5274–5278.
Drevets WC, Gautier C, Price JC, Kupfer DJ, Kinahan PE, Grace AA et al (2001). Amphetamine-induced dopamine release in human ventral striatum correlates with euphoria. Biol Psychiatry 49: 81–96.
Duncan E, Boshoven W, Harenski K, Fiallos A, Tracy H, Jovanovic T et al (2007). An fMRI study of the interaction of stress and cocaine cues on cocaine craving in cocaine-dependent men. Am J Addict 16: 174–182.
Dyr W, Kostowski W (1995). Evidence that the amygdala is involved in the inhibitory effects of 5-HT3 receptor antagonists on alcohol drinking in rats. Alcohol 12: 387–391.
Edwards S, Graham DL, Bachtell RK, Self DW (2007). Region-specific tolerance to cocaine-regulated cAMP-dependent protein phosphorylation following chronic self-administration. Eur J Neurosci 25: 2201–2213.
Everitt BJ, Belin D, Economidou D, Pelloux Y, Dalley JW, Robbins TW (2008). Review. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Phil Trans Royal Soc London B Biol Sci 363: 3125–3135.
Everitt BJ, Robbins TW (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci 8: 1481–1489 (erratum: 9(7): 979).
Everitt BJ, Wolf ME (2002). Psychomotor stimulant addiction: a neural systems perspective. J Neurosci 22: 3312–3320 (erratum: 22(16): 1a).
Ferrario CR, Gorny G, Crombag HS, Li Y, Kolb B, Robinson TE (2005). Neural and behavioral plasticity associated with the transition from controlled to escalated cocaine use. Biol Psychiatry 58: 751–759.
Fowler JS, Volkow ND, Logan J, Alexoff D, Telang F, Wang GJ et al (2008). Fast uptake and long-lasting binding of methamphetamine in the human brain: comparison with cocaine. Neuroimage 43: 756–763.
Franklin TR, Wang Z, Wang J, Sciortino N, Harper D, Li Y et al (2007). Limbic activation to cigarette smoking cues independent of nicotine withdrawal: a perfusion fMRI study. Neuropsychopharmacology 32: 2301–2309.
Freeman AS, Meltzer LT, Bunney BS (1985). Firing properties of substantia nigra dopaminergic neurons in freely moving rats. Life Sci 36: 1983–1994.
Funk CK, O’Dell LE, Crawford EF, Koob GF (2006). Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced ethanol selfadministration in withdrawn, ethanol-dependent rats. J Neurosci 26: 11324–11332. This study showed that blockade of CRF receptors in the area of the central nucleus of the amygdala blocks the increased alcohol intake associated with dependence but not alcohol intake in nondependent animals.
Garavan H, Kaufman JN, Hester R (2008). Acute effects of cocaine on the neurobiology of cognitive control. Phil Trans Royal Soc London B Biol Sci 363: 3267–3276.
George O, Ghozland S, Azar MR, Cottone P, Zorrilla EP, Parsons LH et al (2007). CRF-CRF1 system activation mediates withdrawal-induced increases in nicotine self-administration in nicotine-dependent rats. Proc Natl Acad Sci USA 104: 17198–17203.
George O, Mandyam CD, Wee S, Koob GF (2008). Extended access to cocaine self-administration produces long-lasting prefrontal cortex-dependent working memory impairments. Neuropsychopharmacology 33: 2474–2482.
Gilpin NW, Koob GF (2008). Overview: neurobiology of alcohol dependence with a focus on motivational mechanisms. Alcohol Res Health 31: 185–195.
Gilpin NW, Misra K, Koob GF (2008). Neuropeptide Y in the central nucleus of the amygdala suppresses dependence-induced increases in alcohol drinking. Pharmacol Biochem Behav 90: 475–480.
Goldstein RZ, Volkow ND (2002). Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 159: 1642–1652.
Gonzalez D, Riba J, Bouso JC, Gomez-Jarabo G, Barbanoj MJ (2006). Pattern of use and subjective effects of Salvia divinorum among recreational users. Drug Alcohol Depend 85: 157–162.
Grace AA (2000). The tonic/phasic model of dopamine system regulation and its implications for understanding alcohol and psychostimulant craving. Addiction 95(Suppl 2): S119–S128.
Grant BF, Dawson DA (1998). Age of onset of drug use and its association with DSM-IV drug abuse and dependence: results from the National Longitudinal Alcohol Epidemiologic Survey. J Subst Abuse 10: 163–173.
Grant BF, Dawson DA, Stinson FS, Chou SP, Dufour MC, Pickering RP (2004). The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991–1992 and 2001–2002. Drug Alcohol Depend 74: 223–234.
Grant S, London ED, Newlin DB, Villemagne VL, Liu X, Contoreggi C et al (1996). Activation of memory circuits during cue-elicited cocaine craving. Proc Natl Acad Sci USA 93: 12040–12045.
Greenwell TN, Funk CK, Cottone P, Richardson HN, Chen SA, Rice K et al (2009). Corticotropin-releasing factor-1 receptor antagonists decrease heroin selfadministration in long-, but not short-access rats. Addict Biol 14: 130–143.
Hand TH, Koob GF, Stinus L, Le Moal M (1988). Aversive properties of opiate receptor blockade: evidence for exclusively central mediation in naive and morphine-dependent rats. Brain Res 474: 364–368.
Hebb DO (1972). Textbook of Psychology 3rd edn. WB Saunders: Philadelphia. Heilig M, Koob GF (2007). A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci 30: 399–406.
Heilig M, Koob GF, Ekman R, Britton KT (1994). Corticotropin-releasing factor and neuropeptide Y: role in emotional integration. Trends Neurosci 17: 80–85.
Heimer L, Alheid G (1991). Piecing together the puzzle of basal forebrain anatomy. In: Napier TC, Kalivas PW, Hanin I (eds). The Basal Forebrain: Anatomy to Function (series title: Advances in Experimental Medicine and Biology, vol 295).
Plenum Press: New York. pp 1–42. Heinrichs SC, Menzaghi F, Schulteis G, Koob GF, Stinus L (1995). Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav Pharmacol 6: 74–80.
Heinz A, Siessmeier T, Wrase J, Hermann D, Klein S, Grusser SM et al (2004). Correlation between dopamine D(2) receptors in the ventral striatum and central processing of alcohol cues and craving. Am J Psychiatry 161: 1783–1789 (erratum: 161: 2344).
Hernandez G, Hamdani S, Rajabi H, Conover K, Stewart J, Arvanitogiannis A et al (2006). Prolonged rewarding stimulation of the rat medial forebrain bundle: neurochemical and behavioral consequences. Behav Neurosci 120: 888–904.
Heyser CJ, Roberts AJ, Schulteis G, Koob GF (1999). Central administration of an opiate antagonist decreases oral ethanol self-administration in rats. Alcohol Clin Exp Res 23: 1468–1476.
Hill RT (1970). Facilitation of conditioned reinforcement as a mechanism of psychomotor stimulation. In: Cost E, Garattini S (eds). Amphetamines and Related Compounds. Raven Press: New York. pp 781–795.
Holland PC, Gallagher M (2004). Amygdala–frontal interactions and reward expectancy. Curr Opin Neurobiol 14: 148–155.
Hubner CB, Koob GF (1990). The ventral pallidum plays a role in mediating cocaine and heroin self-administration in the rat. Brain Res 508: 20–29.
Hyman SE, Malenka RC, Nestler EJ (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598.
Hyytia P, Koob GF (1995). GABA-A receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur J Pharmacol 283: 151–159.
Ito R, Dalley JW, Robbins TW, Everitt BJ (2002). Dopamine release in the dorsal striatum during cocaine-seeking behavior under the control of a drug-associated cue. J Neurosci 22: 6247–6253.
Jentsch JD, Olausson P, de la Garza II R, Taylor JR (2002). Impairments of reversal learning and response perseveration after repeated, intermittent cocaine administrations to monkeys. Neuropsychopharmacology 26: 183–190.
Jones S, Bonci A (2005). Synaptic plasticity and drug addiction. Curr Opin Pharmacol 5: 20–25.
June HL, Foster KL, McKay PF, Seyoum R, Woods JE, Harvey SC et al (2003). The reinforcing properties of alcohol are mediated by GABA(A1) receptors in the ventral pallidum. Neuropsychopharmacology 28: 2124–2137.
Justinova Z, Tanda G, Redhi GH, Goldberg SR (2003). Self-administration of delta9- tetrahydrocannabinol (THC) by drug naive squirrel monkeys. Psychopharmacology 169: 135–140.
Kalivas PW (2004). Glutamate systems in cocaine addiction. Curr Opin Pharmacol 4: 23–29.
Kalivas PW, O’Brien C (2008). Drug addiction as a pathology of staged neuroplasticity. Neuropsychopharmacology 33: 166–180.
Kalivas PW, Volkow ND (2005). The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry 162: 1403–1413.
Kelly PH, Iversen SD (1976). Selective 6-OHDA-induced destruction of mesolimbic dopamine neurons: abolition of psychostimulant-induced locomotor activity in rats. Eur J Pharmacol 40: 45–56.
Kenny PJ, Chen SA, Kitamura O, Markou A, Koob GF (2006). Conditioned withdrawal drives heroin consumption and decreases reward sensitivity. J Neurosci 26: 5894–5900.
Kilts CD, Schweitzer JB, Quinn CK, Gross RE, Faber TL, Muhammad F et al (2001). Neural activity related to drug craving in cocaine addiction. Arch Gen Psychiatry 58: 334–341.
Kitamura O, Wee S, Specio SE, Koob GF, Pulvirenti L (2006). Escalation of methamphetamine self-administration in rats: a dose–effect function. Psychopharmacology 186: 48–53.
Koob GF (1992). Drugs of abuse: anatomy, pharmacology, and function of reward pathways. Trends Pharmacol Sci 13: 177–184.
Koob GF (2004). Allostatic view of motivation: implications for psychopathology. In: Bevins RA, Bardo MT (eds). Motivational Factors in the Etiology of Drug Abuse (series title: Nebraska Symposium on Motivation, vol 50). University of Nebraska Press: Lincoln, NE. pp 1–18.
Koob GF (2005). The neurocircuitry of addiction: implications for treatment. Clin Neurosci Res 5: 89–101.
Koob GF (2008). A role for brain stress systems in addiction. Neuron 59: 11–34.
Koob GF (2009a). Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology 56(Suppl 1): 18–31.
Koob GF (2009b). Brain stress systems in the amygdala and addiction. Brain Res (in press).
Koob GF, Everitt BJ, Robbins TW (2008a). Reward, motivation, and addiction. In: Squire LG, Berg D, Bloom FE, Du Lac S, Ghosh A, Spitzer N (eds). Fundamental Neuroscience 3rd edn. Academic Press: Amsterdam. pp 987–1016.
Koob GF, Kandel D, Volkow ND (2008b). Pathophysiology of addiction. In: Tasman A, Kay J, Lieberman JA, First MB, Maj M (eds). Psychiatry 3rd edn, vol 1 Wiley: Chichester. pp 354–378.
Koob GF, Kreek MJ (2007). Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry 164: 1149–1159.
Koob GF, Le Moal M (1997). Drug abuse: hedonic homeostatic dysregulation. Science 278: 52–58. This theoretical review argued that drug addiction involves decreased hedonic homeostatic dysregulation (dysregulation of reward function) driven by both decreased activity in reward pathways and recruitment of brain stress systems.
Koob GF, Le Moal M (2001). Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24: 97–129.
Koob GF, Le Moal M (2005). Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci 8: 1442–1444.
Koob GF, Le Moal M (2006). Neurobiology of Addiction. Academic Press: London.
Koob GF, Le Moal M (2008). Addiction and the brain antireward system. Annu Rev Psychol 59: 29–53.
Koob GF, Lloyd GK, Mason BJ (2009). Development of pharmacotherapies for drug addiction: a Rosetta Stone approach. Nat Rev Drug Discov 8: 500–515.
Koob GF, Nestler EJ (1997). The neurobiology of drug addiction. J Neuropsychiatry Clin Neurosci 9: 482–497.
Kornetsky C, Bain G (1990). Brain-stimulation reward: a model for drug induced euphoria. In: Adler MW, Cowan A (eds). Testing and Evaluation of Drugs of Abuse (series title: Modern Methods in Pharmacology, vol 6). Wiley-Liss: New York. pp 211–231.
Kornetsky C, Esposito RU (1979). Euphorigenic drugs: effects on the reward pathways of the brain. Fed Proc 38: 2473–2476.
Kourrich S, Rothwell PE, Klug JR, Thomas MJ (2007). Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci 27: 7921–7928.
Kufahl P, Li Z, Risinger R, Rainey C, Piacentine L, Wu G et al (2008). Expectation modulates human brain responses to acute cocaine: a functional magnetic resonance imaging study. Biol Psychiatry 63: 222–230.
Langleben DD, Ruparel K, Elman I, Busch-Winokur S, Pratiwadi R, Loughead J et al (2008). Acute effect of methadone maintenance dose on brain FMRI response to heroin-related cues. Am J Psychiatry 165: 390–394.
LaRowe SD, Myrick H, Hedden S, Mardikian P, Saladin M, McRae A et al (2007). Is cocaine desire reduced by N-acetylcysteine? Am J Psychiatry 164: 1115–1117.
Laviolette SR, Alexson TO, van der Kooy D (2002). Lesions of the tegmental pedunculopontine nucleus block the rewarding effects and reveal the aversive effects of nicotine in the ventral tegmental area. J Neurosci 22: 8653–8660.
Le Doux JE (2000). Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184. Le Moal M, Simon H (1991). Mesocorticolimbic dopaminergic network: functional and regulatory roles. Physiol Rev 71: 155–234.
Lee JL (2008). Memory reconsolidation mediates the strengthening of memories by additional learning. Nat Neurosci 11: 1264–1266.
Lee JL, Di Ciano P, Thomas KL, Everitt BJ (2005). Disrupting reconsolidation of drug memories reduces cocaine-seeking behavior. Neuron 47: 795–801.
Li YQ, Li FQ, Wang XY, Wu P, Zhao M, Xu CM et al (2008). Central amygdala extracellular signal-regulated kinase signaling pathway is critical to incubation of opiate craving. J Neurosci 28: 13248–13257.
Logan GD, Schachar RJ, Tannock R (1997). Impulsivity and inhibitory control. Psychol Sci 8: 60–64.
Lu L, Koya E, Zhai H, Hope BT, Shaham Y (2006). Role of ERK in cocaine addiction. Trends Neurosci 29: 695–703.
Madayag A, Lobner D, Kau KS, Mantsch JR, Abdulhameed O, Hearing M et al (2007). Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J Neurosci 27: 13968–13976.
Maisonneuve IM, Ho A, Kreek MJ (1995). Chronic administration of a cocaine ‘binge’ alters basal extracellular levels in male rats: an in vivo microdialysis study. J Pharmacol Exp Ther 272: 652–657.
Mameli-Engvall M, Evrard A, Pons S, Maskos U, Svensson TH, Changeux JP et al (2006). Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron 50: 911–921.
Markou A, Kosten TR, Koob GF (1998). Neurobiological similarities in depression and drug dependence: a self-medication hypothesis. Neuropsychopharmacology 18: 135–174.
Martinez D, Broft A, Foltin RW, Slifstein M, Hwang DR, Huang Y et al (2004). Cocaine dependence and d2 receptor availability in the functional subdivisions of the striatum: relationship with cocaine-seeking behavior. Neuropsychopharmacology 29: 1190–1202 (erratum: 29: 1763).
Martinez D, Gil R, Slifstein M, Hwang DR, Huang Y, Perez A et al (2005). Alcohol dependence is associated with blunted dopamine transmission in the ventral striatum. Biol Psychiatry 58: 779–786.
Martinez D, Narendran R, Foltin RW, Slifstein M, Hwang DR, Broft A et al (2007). Amphetamine-induced dopamine release: markedly blunted in cocaine dependence and predictive of the choice to self-administer cocaine. Am J Psychiatry 164: 622–629.
McBride WJ, Murphy JM, Ikemoto S (1999). Localization of brain reinforcement mechanisms: intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res 101: 129–152.
McClernon FJ, Kozink RV, Lutz AM, Rose JE (2009). 24-h smoking abstinence potentiates fMRI-BOLD activation to smoking cues in cerebral cortex and dorsal striatum. Psychopharmacology 204: 25–35.
McClung CA, Ulery PG, Perrotti LI, Zachariou V, Berton O, Nestler EJ (2004). DeltaFosB: a molecular switch for long-term adaptation in the brain. Mol Brain Res 132: 146–154.
McFarland K, Kalivas PW (2001). The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 21: 8655–8663. This study established a key role of the dorsal frontal cortex-nucleus accumbens-ventral pallidal circuit in cocaine-induced reinstatement.
McFarland K, Lapish CC, Kalivas PW (2003). Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci 23: 3531–3537.
McGregor A, Roberts DCS (1993). Dopaminergic antagonism within the nucleus accumbens or the amygdala produces differential effects on intravenous cocaine self-administration under fixed and progressive ratio schedules of reinforcement. Brain Res 624: 245–252.
Melendez RI, Rodd ZA, McBride WJ, Murphy JM (2004). Involvement of the mesopallidal dopamine system in ethanol reinforcement. Alcohol 32: 137–144.
Melis M, Spiga S, Diana M (2005). The dopamine hypothesis of drug addiction: hypodopaminergic state. Int Rev Neurobiol 63: 101–154.
Milton AL, Lee JL, Everitt BJ (2008). Reconsolidation of appetitive memories for both natural and drug reinforcement is dependent on b-adrenergic receptors. Learn Mem 15: 88–92.
Miranda MI, LaLumiere RT, Buen TV, Bermudez-Rattoni F, McGaugh JL (2003). Blockade of noradrenergic receptors in the basolateral amygdala impairs taste memory. Eur J Neurosci 18: 2605–2610.
Moeller FG, Barratt ES, Dougherty DM, Schmitz JM, Swann AC (2001). Psychiatric aspects of impulsivity. Am J Psychiatry 158: 1783–1793.
Moller C, Wiklund L, Sommer W, Thorsell A, Heilig M (1997). Decreased experimental anxiety and voluntary ethanol consumption in rats following central but not basolateral amygdala lesions. Brain Res 760: 94–101.
Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A et al (2009). N-acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci 12: 182–189.
Murray EA (2007). The amygdala, reward and emotion. Trends Cogn Sci 11: 489–497.
Naqvi NH, Bechara A (2009). The hidden island of addiction: the insula. Trends Neurosci 32: 56–67.
Naqvi NH, Rudrauf D, Damasio H, Bechara A (2007). Damage to the insula disrupts addiction to cigarette smoking. Science 315: 531–534. This study showed that damage to the insula in human smokers was associated with cessation of smoking, establishing a link between the insula and nicotine addiction.
Nauta JH, Haymaker W (1969). Hypothalamic nuclei and fiber connections. In: Haymaker W, Anderson E, Nauta WJH (eds). The Hypothalamus. Charles C Thomas: Springfield, IL. pp 136–209.
Nelson CL, Milovanovic M, Wetter JB, Ford KA, Wolf ME (2009). Behavioral sensitization to amphetamine is not accompanied by changes in glutamate receptor surface expression in the rat nucleus accumbens. J Neurochem 109: 35–51.
Nestler EJ (2005). Is there a common molecular pathway for addiction? Nat Neurosci 8: 1445–1449. This review summarizes a body of work characterizing the role of molecular changes mediating the transition from drug taking to addiction with a special emphasis on the accumulation of the transcription factor DFosB in the nucleus accumbens following chronic drug exposure.
Neugebauer V, Li W, Bird GC, Han JS (2004). The amygdala and persistent pain. Neuroscientist 10: 221–234.
O’Dell LE, Koob GF (2007). Nicotine deprivation effect in rats with intermittent 23-h access to intravenous nicotine self-administration. Pharmacol Biochem Behav 86: 346–353.
Olds J, Milner P (1954). Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol 47: 419–427.
Orsini C, Koob GF, Pulvirenti L (2001). Dopamine partial agonist reverses amphetamine withdrawal in rats. Neuropsychopharmacology 25: 789–792.
Pierce RC, Bell K, Duffy P, Kalivas PW (1996). Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci 16: 1550–1560.
Pulvirenti L, Koob GF (1993). Lisuride reduces psychomotor retardation during withdrawal from chronic intravenous amphetamine self-administration in rats. Neuropsychopharmacology 8: 213–218.
Rachlin H, Green L (1972). Commitment, choice and self-control. J Exp Anal Behav 17: 15–22.
Robbins TW (1976). Relationship between reward-enhancing and stereotypical effects of psychomotor stimulant drugs. Nature 264: 57–59.
Roberts AJ, Heyser CJ, Cole M, Griffin P, Koob GF (2000). Excessive etanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology 22: 581–594.
Roberts DCS (1992). Neural substrates mediating cocaine reinforcement: the role of monoamine systems. In: Lakoski JM, Galloway MP, White FJ (eds). Cocaine: Pharmacology, Physiology and Clinical Strategies. CRC Press: Boca Raton, FL. pp 73–90.
Robinson TE, Berridge KC (1993). The neural basis of drug craving: an incentivesensitization theory of addiction. Brain Res Rev 18: 247–291.
Robledo P, Koob GF (1993). Two discrete nucleus accumbens projection áreas differentially mediate cocaine self-administration in the rat. Behav Brain Res 55: 159–166.
Rocha BA, Fumagalli F, Gainetdinov RR, Jones SR, Ator R, Giros B et al (1998). Cocaine self-administration in dopamine-transporter knockout mice. Nat Neurosci 1: 132–137.
Rossetti ZL, Hmaidan Y, Gessa GL (1992). Marked inhibition of mesolimbic dopamine release: a common feature of ethanol, morphine, cocaine and amphetamine abstinence in rats. Eur J Pharmacol 221: 227–234.
Russell MAH (1976). What is dependence?. In: Edwards G (ed). Drugs and Drug Dependence. Lexington Books: Lexington, MA. pp 182–187.
Saal D, Dong Y, Bonci A, Malenka RC (2003). Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582 (erratum: 38: 359).
Salamone JD, Correa M, Farrar A, Mingote SM (2007). Effort-related functions of nucleus accumbens dopamine and associated forebrain circuits. Psychopharmacology 191: 461–482.
Sanchis-Segura C, Spanagel R (2006). Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol 11: 2–38.
Sarnyai Z, Biro E, Gardi J, Vecsernyes M, Julesz J, Telegdy G (1995). Brain corticotropin-releasing factor mediates ‘anxiety-like’ behavior induced by cocaine withdrawal in rats. Brain Res 675: 89–97.
Schoenbaum G, Saddoris MP, Ramus SJ, Shaham Y, Setlow B (2004). Cocaine experienced rats exhibit learning deficits in a task sensitive to orbitofrontal cortex lesions. Eur J Neurosci 19: 1997–2002.
Schulteis G, Ahmed SH, Morse AC, Koob GF, Everitt BJ (2000). Conditioning and opiate withdrawal: the amygdala links neutral stimuli with the agony of overcoming drug addiction. Nature 405: 1013–1014.
Schulteis G, Stinus L, Risbrough VB, Koob GF (1998). Clonidine blocks acquisition but not expression of conditioned opiate withdrawal in rats. Neuropsychopharmacology 19: 406–416.
Schultz W (2007). Multiple dopamine functions at different time courses. Annu Rev Neurosci 30: 259–288.
Shaham Y, Shalev U, Lu L, de Wit H, Stewart J (2003). The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology 168: 3–20.
Shalev U, Grimm JW, Shaham Y (2002). Neurobiology of relapse to heroin and cocaine seeking: a review. Pharmacol Rev 54: 1–42. This review summarizes the neurocircuitry associated with drug-, cue-, and stress-induced relapse determined from extensive work with animal models.
Sharma A, Brody AL (2009). In vivo brain imaging of human exposure to nicotine and tobacco. Handb Exp Pharmacol 192: 145–171.
Shaw-Lutchman TZ, Barrot M, Wallace T, Gilden L, Zachariou V, Impey S et al (2002). Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci 22: 3663–3672.
Solomon RL, Corbit JD (1974). An opponent-process theory of motivation: 1. Temporal dynamics of affect. Psychol Rev 81: 119–145.
Stein L (1962). Effects and interactions of imipramine, chlorpromazine, reserpine, and amphetamine on self-stimulation: possible neurophysiological basis of depression. Recent Adv Biol Psychiatry 4: 288–309.
Stinus L, Cador M, Zorrilla EP, Koob GF (2005). Buprenorphine and a CRF1 antagonist block the acquisition of opiate withdrawal-induced conditioned place aversion in rats. Neuropsychopharmacology 30: 90–98.
Substance Abuse and Mental Health Services Administration (2008). Results from the 2007 National Survey on Drug Use and Health: National Findings (Office of Applied Statistics, NSDUH Series H-34, DHHS Publication No. SMA 08-4343). Rockville, MD.
Sutton MA, Schmidt EF, Choi KH, Schad CA, Whisler K, Simmons D et al (2003). Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature 421: 70–75.
Tanda G, Pontieri FE, Di Chiara G (1997). Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common m1 opioid receptor mechanism. Science 276: 2048–2050.
Thorsell A, Rapunte-Canonigo V, O’Dell L, Chen SA, King A, Lekic D et al (2007). Viral vector-induced amygdala NPY overexpression reverses increased alcohol intake caused by repeated deprivations in Wistar rats. Brain 130: 1330–1337.
Thorsell A, Slawecki CJ, Ehlers CL (2005a). Effects of neuropeptide Y and corticotropin-releasing factor on ethanol intake in Wistar rats: interaction with chronic ethanol exposure. Behav Brain Res 161: 133–140.
Thorsell A, Slawecki CJ, Ehlers CL (2005b). Effects of neuropeptide Y on appetitive and consummatory behaviors associated with alcohol drinking in Wistar rats with a history of ethanol exposure. Alcohol Clin Exp Res 29: 584–590.
Tiffany ST, Carter BL, Singleton EG (2000). Challenges in the manipulation, assessment and interpretation of craving relevant variables. Addiction 95(Suppl 2): s177–s187.
Todtenkopf MS, Parsegian A, Naydenov A, Neve RL, Konradi C, Carlezon Jr WA (2006). Brain reward regulated by AMPA receptor subunits in nucleus accumbens shell. J Neurosci 26: 11665–11669.
Tomasi D, Goldstein RZ, Telang F, Maloney T, Alia-Klein N, Caparelli EC et al (2007a). Widespread disruption in brain activation patterns to a working memory task during cocaine abstinence. Brain Res 1171: 83–92.
Tomasi D, Goldstein RZ, Telang F, Maloney T, Alia-Klein N, Caparelli EC et al (2007b). Thalamo-cortical dysfunction in cocaine abusers: implications in attention and perception. Psychiatry Res 155: 189–201.
Tornatzky W, Miczek KA (2000). Cocaine self-administration ‘binges’: transition from behavioral and autonomic regulation toward homeostatic dysregulation in rats. Psychopharmacology 148: 289–298.
Tucci S, Cheeta S, Seth P, File SE (2003). Corticotropin releasing factor antagonist, a-helical CRF941, reverses nicotine-induced conditioned, but not unconditioned, anxiety. Psychopharmacology 167: 251–256.
Tzschentke TM (1998). Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol 56: 613–672.
Ungless MA, Whistler JL, Malenka RC, Bonci A (2001). Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583–587.
Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP et al (2002). Increased ethanol self-administration and anxiety-like behavior during acute withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res 26: 1494–1501.
Vanderschuren LJ, Everitt BJ (2004). Drug seeking becomes compulsive after prolonged cocaine self-administration. Science 305: 1017–1019.
Vezina P (2004). Sensitization of midbrain dopamine neuron reactivity and the self-administration of psychomotor stimulant drugs. Neurosci Biobehav Rev 27: 827–839.
Volkow ND, Chang L, Wang GJ, Fowler JS, Ding YS, Sedler M et al (2001a). Low level of brain dopamine D2 receptors in methamphetamine abusers: association with metabolism in the orbitofrontal cortex. Am J Psychiatry 158: 2015–2021. This study shows an association between the decreases in dopamine function in addiction and decreased function of the orbitofrontal cortex, establishing a key link between compromised striatal activity and orbitofrontal dysfunction in addiction.
Volkow ND, Fowler JS, Wang GJ, Ding YS, Gatley SJ (2002). Role of dopamine in the therapeutic and reinforcing effects of methylphenidate in humans: results from imaging studies. Eur Neuropsychopharmacol 12: 557–566.
Volkow ND, Fowler JS, Wang GJ, Hitzemann R, Logan J, Schlyer DJ et al (1993). Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse 14: 169–177.
Volkow ND, Fowler JS, Wang GJ (2004a). The addicted human brain viewed in the light of imaging studies: brain circuits and treatment strategies. Neuropharmacology 47(Suppl 1): 3–13.
Volkow ND, Fowler JS, Wang GJ, Swanson JM (2004b). Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry 9: 557–569.
Volkow ND, Gillespie H, Mullani N, Tancredi L, Grant C, Valentine A et al (1996a). Brain glucose metabolism in chronic marijuana users at baseline and during marijuana intoxication. Psychiatry Res 67: 29–38.
Volkow ND, Swanson JM (2003). Variables that affect the clinical use and abuse of methylphenidate in the treatment of ADHD. Am J Psychiatry 160: 1909–1918.
Volkow ND, Wang G, Fowler JS, Logan J, Gerasimov M, Maynard L et al (2001b). Therapeutic doses of oral methylphenidate significantly increase extracelular dopamine in the human brain. J Neurosci 21: RC121.
Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN et al (1997a). Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature 386: 827–830.
Volkow ND, Wang GJ, Fowler JS, Franceschi D, Thanos PK, Wong C et al (2000). Cocaine abusers show a blunted response to alcohol intoxication in limbic brain regions. Life Sci 66: PL161–PL167.
Volkow ND, Wang GJ, Fowler JS, Gatley SJ, Ding YS, Logan J et al (1996b). Relationship between psychostimulant-induced ‘high’ and dopamine transporter occupancy. Proc Natl Acad Sci USA 93: 10388–10392.
Volkow ND, Wang GJ, Fowler JS, Hitzemann R, Angrist B, Gatley SJ et al (1999). Association of methylphenidate-induced craving with changes in right striatoorbitofrontal metabolism in cocaine abusers: implications in addiction. Am J Psychiatry 156: 19–26.
Volkow ND, Wang GJ, Fowler JS, Hitzemann R, Gatley SJ, Dewey SS et al (1998). Enhanced sensitivity to benzodiazepines in active cocaine-abusing subjects: a PET study. Am J Psychiatry 155: 200–206.
Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, Hitzemann R et al (1997b). Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature 386: 830–833. This study using PET showed decreased release of dopamine in the striatum and a decreased ‘high’ produced by methylphenidate, suggesting a compromised striatal dopamine system in addiction.
Volkow ND, Wang GJ, Ma Y, Fowler JS, Wong C, Ding YS et al (2005). Activation of orbital and medial prefrontal cortex by methylphenidate in cocaine-addicted subjects but not in controls: relevance to addiction. J Neurosci 25: 3932–3939.
Volkow ND, Wang GJ, Ma Y, Fowler JS, Zhu W, Maynard L et al (2003). Expectation enhances the regional brain metabolic and the reinforcing effects of stimulants in cocaine abusers. J Neurosci 23: 11461–11468.
Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Childress AR et al (2006). Cocaine cues and dopamine in dorsal striatum: mechanism of craving in cocaine addiction. J Neurosci 26: 6583–6588.
Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Childress AR et al (2008a). Dopamine increases in striatum do not elicit craving in cocaine abusers unless they are coupled with cocaine cues. Neuroimage 39: 1266–1273.
Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Jayne M et al (2007). Profound decreases in dopamine release in striatum in detoxified alcoholics: posible orbitofrontal involvement. J Neurosci 27: 12700–12706.
Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Wong C et al (2008b). Sleep deprivation decreases binding of [11C]raclopride to dopamine D2/D3 receptors in the human brain. J Neurosci 28: 8454–8461.
Wang GJ, Volkow ND, Fowler JS, Logan J, Abumrad NN, Hitzemann RJ et al (1997). Dopamine D2 receptor availability in opiate-dependent subjects before and after naloxone-precipitated withdrawal. Neuropsychopharmacology 16: 174–182.
Wang Z, Faith M, Patterson F, Tang K, Kerrin K, Wileyto EP et al (2007). Neural substrates of abstinence-induced cigarette cravings in chronic smokers. J Neurosci 27: 14035–14040.
Watkins SS, Stinus L, Koob GF, Markou A (2000). Reward and somatic changes during precipitated nicotine withdrawal in rats: centrally and peripherally mediated effects. J Pharmacol Exp Ther 292: 1053–1064.
Wee S, Wang Z, Woolverton WL, Pulvirenti L, Koob GF (2007). Effect of aripiprazole, a partial D2 receptor agonist, on increased rate of methamphetamine selfadministration in rats with prolonged access. Neuropsychopharmacology 32: 2238–2247.
Wee S, Orio L, Ghirmai S, Cashman J, Koob GF (2009). Inhibition of kappa opioid receptors attenuates the increased motivation for cocaine in rats with extended access to cocaine. Psychopharmacology (in press).
Weiss F, Ciccocioppo R, Parsons LH, Katner S, Liu X, Zorrilla EP et al (2001). Compulsive drug-seeking behavior and relapse: neuroadaptation, stress, and conditioning factors. In: Quinones-Jenab V (ed). The Biological Basis of Cocaine Addiction (series title: Annals of the New York Academy of Sciences, vol 937) New York Academy of Sciences: New York. pp 1–26.
Weiss F, Markou A, Lorang MT, Koob GF (1992). Basal extracellular dopamine levels in the nucleus accumbens are decreased during cocaine withdrawal after unlimited-access self-administration. Brain Res 593: 314–318.
Weiss F, Parsons LH, Schulteis G, Hyytia P, Lorang MT, Bloom FE et al (1996). Ethanol self-administration restores withdrawal-associated deficiencies in accumbal dopamine and 5-hydroxytryptamine release in dependent rats. J Neurosci 16: 3474–3485.
White NM (1996). Addictive drugs as reinforcers: multiple partial actions on memory systems. Addiction 91: 921–949.
Whitelaw RB, Markou A, Robbins TW, Everitt BJ (1996). Excitotoxic lesions of the basolateral amygdala impair the acquisition of cocaine-seeking behaviour under a second-order schedule of reinforcement. Psychopharmacology 127: 213–224.
Wikler A (1952). A psychodynamic study of a patient during experimental selfregulated re-addiction to morphine. Psychiatr Q 26: 270–293.
Wise RA (1978). Catecholamine theories of reward: a critical review. Brain Res 152: 215–247.
Wolf ME (2002). Addiction: making the connection between behavioral changes and neuronal plasticity in specific pathways. Mol Intervent 2: 146–157.
Wolf ME, Sun X, Mangiavacchi S, Chao SZ (2004). Psychomotor stimulants and neuronal plasticity. Neuropharmacology 47(Suppl 1): 61–79.
Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM et al (2004). Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41: 625–638.
Yeomans J, Baptista M (1997). Both nicotinic and muscarinic receptors in ventral tegmental area contribute to brain-stimulation reward. Pharmacol Biochem Behav 57: 915–921.
Yin HH, Ostlund SB, Knowlton BJ, Balleine BW (2005). The role of the dorsomedial striatum in instrumental conditioning. Eur J Neurosci 22: 513–523.
Zubieta JK, Gorelick DA, Stauffer R, Ravert HT, Dannals RF, Frost JJ (1996). Increased mu opioid receptor binding detected by PET in cocaine-dependent men is associated with cocaine craving. Nat Med 2: 1225–1229.
Zubieta J, Lombardi U, Minoshima S, Guthrie S, Ni L, Ohl LE et al (2001). Regional cerebral blood flow effects of nicotine in overnight abstinent smokers. Biol Psychiatry 49: 906–913.
La investigación sobre la adicción explora muchos aspectos de cómo y por qué se desarrolla esta enfermedad. Aquí están cuatro de las preguntas más difíciles.
1. ¿En qué medida debemos definir la adicción?
Por qué es importante. A menudo pensamos en la adicción en relación con el uso indebido de sustancias como el alcohol, la heroína y la cocaína. ¿Pero los comportamientos compulsivos que involucran sexo, videojuegos y juegos de apuestas también deben tratarse como adicciones?
Lo que sabemos es que el juego es la única adicción conductual reconocida oficialmente. Pero hay cada vez más pruebas de que otros comportamientos se asemejan al uso indebido de sustancias en su neurobiología subyacente y a la forma en que responden al tratamiento (S62).
Próximos pasos. Los investigadores desean pasar de un sistema que clasifica la adicción según los síntomas clínicos a uno más arraigado en una comprensión mecánica de la enfermedad, con un mayor papel para la imagen genética y la ciencia cognitiva.
2. ¿Cómo se ve afectada la adicción por nuestros genes?
Por qué es importante. Una mejor comprensión de los factores genéticos que influyen en el riesgo de adicción podría revolucionar la forma en que se diagnostica, trata y previene la adicción.
Lo que sabemos. Numerosos estudios de adopción y estudios de gemelos han demostrado que el riesgo de adicción es hereditario en un 50%. Pero la identificación de los factores genéticos y la medida en que interactúan entre sí y con el medio ambiente ha sido más complicada (S48).
Próximos pasos. Los investigadores están buscando con mayor detalle las posibles fuentes de variaciones genéticas (como el número de copias de los genes o la existencia de genes raros) y los cambios epigenéticos en la forma en que se expresan los genes.
3. Si la adicción cambia las conexiones en el cerebro, ¿podemos reconexionar el circuito?
Por qué es importante. Los científicos saben que el uso de sustancias adictivas provoca cambios físicos en el cerebro que pueden conducir a la adicción. Lo que recién comienzan a entender es cómo se pueden revertir esos cambios para tratar la enfermedad.
Lo que sabemos. Al utilizar la cirugía cerebral guiada por optogenética en ratones, los investigadores han podido identificar un tipo de receptor de dopamina que parece tener un papel crucial en la adicción. El bloqueo de este receptor ha revertido los síntomas de la adicción a la cocaína en ratones (S50).
Próximos pasos. Por razones prácticas y éticas, los métodos optogenéticos no se pueden usar en humanos, pero su uso creciente en el laboratorio podría acelerar el descubrimiento de medicamentos para atacar y restablecer los circuitos de recompensa que están sobrecargados en la adicción.
4. ¿Podríamos pasar desde el programa de recuperación de 12 pasos a una sola toma?
Por qué es importante. Uno de los mayores obstáculos en el tratamiento de la adicción es prevenir las recaídas: las personas pueden caer del vagón después de años de abstinencia. Una vacuna que neutraliza una sustancia antes de que llegue al cerebro podría evitar que las personas regresen a los viejos hábitos.
Lo que sabemos. Los investigadores han desarrollado vacunas antidrogas durante más de dos décadas y tienen un candidato que engaña a los sistemas inmunitarios de las ratas para que piensen que la heroína inyectada es un patógeno, por lo que la droga se neutraliza rápidamente antes de que llegue al cerebro (S53).
Próximos pasos. Sin un ensayo de una vacuna eficaz en humanos, no se puede abordar la preocupación sobre si una vacuna podría llevar a que los usuarios de drogas tomen dosis cada vez más altas, pero es poco probable que las compañías farmacéuticas hagan caja.
Resumen: Este comentario aborda los principales problemas relacionados con el diagnóstico de adicción con sus innumerables manifestaciones, con especial atención a la adicción que involucra el uso de Internet o Adicción a Internet (IA). Destaca las inconsistencias y la lógica equivocada utilizada por la American Psychiatric Association (APA) en el DSM-5, particularmente en lo que respecta a su manejo del fenómeno de las conductas adictivas relacionadas con el uso de Internet. La decisión de la APA de descartar el problema de IA a favor de un diagnóstico fabricado basado en uno de sus subtipos, Internet Trastorno de juego (IGD), se agrega a la confusión en lugar de una orientación adecuada de la evaluación y el tratamiento. Es esencial que los proveedores de atención médica estén más atentos a las necesidades de los pacientes / clientes que tienen adicciones que pueden involucrar sustancias y / u otros comportamientos patológicos, especialmente relacionados con el uso de Internet.
Seguimos preocupados por el Manual de diagnóstico y estadístico de trastornos mentales, versión 5 (DSM-5), que sigue siendo un estándar en evaluaciones psicológicas o psiquiátricas, ya que contiene inconsistencias, contradicciones y la perpetuación del enfoque en la nomenclatura de diagnóstico que está fuera de sintonía con La investigación y la práctica actual, especialmente en el campo de la medicina de la adicción. La revisión de Love, Laier, Brand, Hatch y Hajela (2015) hace un trabajo sólido para ilustrar la investigación sustancial en apoyo a Internet Addiction (IA), así como su subtipo Internet Pornography Addiction (IPA). Sin embargo, creemos que es importante articular de manera más completa la falta de lógica y las inconsistencias demostradas por la APA en el DSM-5 (American Psychiatric Association [APA], 2013). Como tal, aquí ofrecemos algunas ideas para guiar a los proveedores de atención médica en el mantenimiento de la claridad relacionada con la adicción y sus innumerables manifestaciones relacionadas con sustancias y / o comportamientos patológicos, especialmente en relación con diversos aspectos del uso de Internet, donde los juegos y la pornografía son subconjuntos significativamente problemáticos.
Aunque muchos intentan afirmar lo contrario, aprovechando el lenguaje inconsistente de la APA (2013, p. 481) dentro del DSM-5 («Por lo tanto, los grupos de conductas repetitivas, que en algunos términos son adicciones conductuales … no se incluyen porque en este momento … «) La APA reconoce de hecho el fenómeno de las conductas adictivas. Esto también se puede ver a través de múltiples ejemplos que rodean al DSM-5, como la creación de un subcapítulo de «Trastornos no relacionados con sustancias» en el capítulo «Trastornos relacionados con sustancias y adicción». En apoyo adicional del reconocimiento implícito de la APA de la existencia de «adicciones de comportamiento», la APA (2013, p. 481) trasladó el Trastorno de Juego (GD), que antes se llamaba Juego Patológico, al subcapítulo recién formado, basado en su » «evidencia de que las conductas de juego activan sistemas de recompensa similares a los activados por drogas de abuso y producen algunos síntomas de comportamiento que parecen comparables a los producidos por los trastornos por uso de sustancias». Estamos de acuerdo con esta decisión y declaración, como lo demuestra la investigación en neurociencia en el Love et al. (2015), la revisión destaca las características patológicas comunes entre los diversos comportamientos que forman parte de la adicción.
Además, en la introducción al diagnóstico de Trastorno de juegos de Internet (IGD) en la «Sección III: Condiciones para estudio adicional» del DSM-5, la APA (2013, págs. 796–797) se indica en la sección «Características asociadas que respaldan el diagnóstico». Sección de IGD: «Las personas con juegos compulsivos de Internet han demostrado activación cerebral en regiones específicas provocadas por la exposición al juego de Internet, pero no se limitan a recompensar las estructuras del sistema». También estamos de acuerdo con esta afirmación, ya que también está respaldada por grandes cantidades de neurocientíficos Investigaciones ilustradas dentro de Love et al. (2015) Revisión. Sin embargo, la APA hizo la declaración categórica antes mencionada, descontando la existencia de adicciones conductuales. Algo confuso, esa declaración se hizo inmediatamente después de afirmar que algunos comportamientos son adictivos, en particular los juegos de azar, y luego agregar IGD al DSM-5. En conjunto, esto crea una ilógica que plantea la siguiente pregunta: ¿Existen “adicciones conductuales” en el mundo psiquiátrico o no?
APA (2013, p. 481) declaró que «… ‘evidencia insuficiente revisada por pares para establecer los criterios de diagnóstico y las descripciones de los cursos’ fue la razón para el rechazo de ‘adicciones de comportamiento'». Más allá del hecho de que esta declaración indica una prioridad importancia de un proceso de calificación basado en criterios y orientado fenomenológicamente, este parece ser un requisito desconcertante ya que los criterios diagnósticos para GD son esencialmente los mismos que los propuestos para otras conductas adictivas (Goodman, 2001; Griffiths, 2005; Hagedorn, 2009; Potenza, 2006). La declaración en su totalidad es un tanto absurda, dado que los trastornos a los que hace referencia la APA (adicción al sexo, adicción al ejercicio y adicción a las compras) son diagnósticos que nunca se propusieron para su consideración o inclusión en el DSM-5. El uso de esta norma se vuelve particularmente problemático cuando se considera el hecho de que muchos de los criterios para la IGD se tomaron literalmente de los criterios establecidos para la adicción a Internet (IA), con la adición de la palabra «juegos». De lo contrario, el uso de estas normas para excluir IA en favor de IGD parecen, en el mejor de los casos, desinformadas y, en el peor, falsas.
La justificación de favorecer un diagnóstico de IGD sobre IA basado en una «literatura considerable» es igualmente problemática e ilógica. La frase subjetiva «literatura considerable» es imposible de descifrar, ya que no se ofrece un estándar formal. Si bien existe una literatura considerable de estudios acumulados en la IGD, la revisión de Love et al (2015) ha ilustrado que existe una literatura igualmente considerable para la IA en sí, así como sus otros subtipos propuestos. En apoyo de su declaración de «literatura considerable», la APA (2013, p. 796) declaró que el grupo de trabajo del DSM-5 revisó más de 240 artículos sobre el tema de la IGD, encontrando «algunas similitudes de comportamiento de los juegos por Internet con los juegos de azar». el trastorno y el uso de sustancias”. Si bien es cierto que se han publicado cientos de artículos y que se han establecido similitudes entre los juegos de azar por Internet, los juegos de azar y la adicción que involucran sustancias, la precisión general de su investigación afirmada es cuestionable. Esto se debe a la confusión generalizada del problema de IA con su subtipo de IGD. Además de la declaración abierta a este efecto («el trastorno de los juegos de Internet (también conocido como trastorno de uso de Internet, la adicción a Internet o la adicción a los juegos) tiene mérito como un trastorno independiente)» este problema se puede ver en la elección de las referencias presentadas Para apoyar el diagnóstico. De las 14 referencias enumeradas en la sección de IGD, 13 fueron a revistas revisadas por pares (Du et al., 2011; Fu, Chan, Wong, & Yip, 2010; Han, Hwang, & Renshaw, 2010; Kim et al., 2011; Ko, Yen, Chen, Chen, y Yen, 2005; Shek, Tang, & Lo, 2009; Tao et al., 2010; Tsitsika et al., 2011; Van Rooij, Schoenmakers, Vermulst, Van den Eijnden, y Van de Mheen, 2011; Weinstein & Lejoyeur, 2010; Widyanto, Griffiths, & Brunsden, 2011; Yuan et al., 2011; Zhou et al., 2011), y uno fue una referencia a un artículo de la revista de cultura popular Wired) sobre IA en China (Stewart, 2010). Entre los artículos revisados por pares, solo tres artículos están enfocados específicamente en IG (Du et al., 2011; Han et al., 2010; Van Rooij et al., 2011). De los 10 restantes artículos, cuatro estudios se refieren al juego como uno de los tres subtipos de IA (Kim et al., 2011; Shek et al., 2009; Tao et al., 2010; Weinstein y Lejoyeux, 2010; Zhou et al. 2011), uno hace referencia al juego como uno de los diez subtipos (Widyanto et al., 2011), tres hacen uso de los términos «game» y «gaming» y se entrelazan con otros términos relacionados con Internet como «juegos de azar» y «pornografía» (Fu et al., 2010; Tsitsika et al., 2011; Yuan et al., 2011), y dos se refieren al «uso de Internet» generalmente sin subtipos (Fu, Chan, Wong y Yip, 2010; Ko, Yen, Chen, Chen, et al., 2005).
En otra declaración que ilustra su desconexión con la realidad del problema, la APA hizo referencia a un componente social como único y esencial para la IGD: «La característica esencial del trastorno de los juegos de Internet es la participación persistente y recurrente en los juegos de computadora, generalmente juegos de grupo, … actividades que incluyen un aspecto significativo de las interacciones sociales durante el juego. Los aspectos del equipo parecen ser una motivación clave” (APA, 2013a, p. 797). El reconocimiento de la importancia de la interacción social y el refuerzo de variables no es de ninguna manera exclusivo de los juegos de Internet. De hecho, Young enfatizó el cibersexo y las relaciones cibernéticas como dos factores en su lista inicial de subtipos para IA (Young, 1998). Davis (2001) también mencionó al cibersexo como una manifestación del uso problemático específico de Internet (SPIU). Tsitsika et al. (2011) (cuyo estudio fue citado por la APA como referencia específica de IGD) encontró que las redes sociales en línea, los juegos de rol, los juegos de rol y la visualización de pornografía son factores de riesgo para IA. Además, Meerkerk, Eijnden y Garretsen (2006) investigaron 11 actividades potenciales en las que las personas se involucran en Internet. Los autores encontraron que los juegos y la erótica (pornografía) son los usos principales de Internet mediante un análisis transversal. Sin embargo, empleando un análisis longitudinal, la erótica fue el predictor más fuerte de IA. Kim y col. (2011) (otro estudio citado por la APA como referencia específica de IGD) declaró: “Los sujetos de IAD usaban Internet casi todos los días, y pasaban más de 8 horas … todos los días frente al monitor, principalmente para charlar con amigos cibernéticos, jugando juegos en línea y viendo pornografías en línea o películas para adultos». Finalmente, la influencia de la interacción social es inherente al concepto de redes sociales / Facebook (Emre & _I” BPULAN, 2012; Karaiskos, Tzavellas, Balta, & Paparrigopoulos, 2010; Kittinger, Correia, & Irons, 2012; Koc & Gulyagci, 2013; Kuss & Griffiths, 2011; Milo sevi c-Ä orÄ’evi c & Ze zelj, 2014; Rosen, Whaling, Rab, Carrier, y Cheever, 2013; Salehan y Negahban, 2013; Turel, He, Xue, Xiao y Bechara, 2014; Weiss & Samenow, 2010). Por lo tanto, es erróneo utilizar este concepto como un factor de delimitación que separa la adicción al juego de las otras «adicciones del comportamiento» mencionadas anteriormente.
Teniendo en cuenta el hecho de que, aunque la APA reconoció la raíz de los criterios de IGD según se adaptó de los diagnósticos propuestos de Tao et al. (2010) para la DIA, que incluían informalmente tres subtipos de IA y se basaban en la propuesta de Block (2008) para la IA, que incluyó formalmente los mismos tres subtipos (juego excesivo, preocupaciones sexuales y mensajes de correo electrónico / texto; posteriormente revisado a Redes sociales; Potenza, 2014). Es paradójico e irónico que la APA abrazara los juegos, pero excluyera explícitamente los otros dos subtipos. Es aún más desconcertante que los criterios presentados para diagnosticar la DCI se basen en gran medida en criterios confiables y validados e instrumentos de evaluación diseñados inicialmente para diagnosticar la EI.
El 1 de diciembre de 2012, los Fideicomisarios de la APA votaron sobre la versión final del DSM-5. IA se propuso formalmente como un nuevo trastorno, pero no se incluyó. En cambio, a puerta cerrada, la APA creó un diagnóstico de IGD, originalmente destinado a ser un subtipo de IA. Este diagnóstico nunca se propuso formalmente y la comunidad profesional no tuvo la oportunidad de proporcionar feedback y comentarios sobre el nuevo diagnóstico. En cuanto a por qué no se incluyó la IA, la totalidad de los factores apunta hacia tres razones posibles.
Se puede especular lógicamente que un argumento representativo para el cambio en el diagnóstico puede haber sido el «argumento del mecanismo de entrega» (Kim & Kim, 2010; King & Delfabbro, 2013; Starcevic, 2013). Este argumento sostiene que Internet es solo un mecanismo de entrega para otras formas de medios, y uno no puede ser adicto a un mecanismo de entrega. Se hizo una analogía de que los alcohólicos no son adictos a las botellas. Esta especulación está respaldada por el hecho de que el diagnóstico más amplio, el desorden de uso de Internet (DIU), se reescribió en el diagnóstico más específico de IGD. Esto coincide con el concepto original de SPIU de Davis (2001), así como con la versión actualizada de Brand, Young y Laier (2014) de Adicción específica a Internet (SIA). Esto también coincide con la diferenciación propuesta por Griffiths entre «adicciones a Internet» y «adicciones en Internet» (Griffiths, King y Demetrovics, 2014).
Sin embargo, el «argumento del mecanismo de administración» podría considerarse discutible, ya que es bien conocido con el uso patológico de sustancias que el uso de agujas en sí mismo es reforzado o «adictivo», por lo que las personas sienten alivio a través del estímulo de usar una aguja, incluso cuando no hay drogas. entregado; y las diferentes rutas de ingestión se convierten en parte de la adicción a medida que la enfermedad avanza, de modo que una persona con adicción puede ser activada al observar el comportamiento de ingestión independientemente de la sustancia que se consume. Sin embargo, si el argumento del mecanismo de entrega fuera la preocupación de la APA, una decisión más directa y lógica hubiera sido retener el diagnóstico propuesto de IA y simplemente requerir subtipos, como juegos, pornografía, redes sociales, compras, etc. exactamente los mismos criterios, referencias y la mayor parte de la redacción actualmente listada para IGD se podría haber mantenido, con solo la palabra «comportamiento» usada en lugar de la palabra «juego». La preocupación de volverse adicto a un mecanismo de entrega se eliminaría, y el progreso científico podría continuar en la amplia gama de comportamientos potencialmente problemáticos relacionados con el uso de Internet. Esto también sería análogo al término general «Trastorno por uso de sustancias», con una delineación posterior de sustancias específicas como el Trastorno por consumo de alcohol, el Trastorno por consumo de cocaína y el Trastorno por consumo de cannabis.
En y sobre el DSM-5, la APA tomó decisiones y declaraciones que no pueden defenderse científicamente y, en cambio, sugieren que las políticas sociales pueden haber sido la base de sus decisiones. Por ejemplo, como Love et al. (2015) ilustrado, la Sociedad Americana de Medicina de la Adicción (ASAM) lanzó una nueva definición formal de adicción altamente científica y muy específica en 2011, que no solo pronunció la adicción como un trastorno médico, sino que también hizo referencia explícita al concepto de comportamiento como adictivo ( ASAM, 2011). Incluso antes de esto, el vicepresidente Joe Biden y la neurocientífica líder en adicciones Nora Volkow propusieron la «Ley de Reconocimiento de la Adicción como una Enfermedad de 2007» en un intento por cambiar el nombre del Instituto Nacional de Abuso de Drogas al Instituto Nacional de Enfermedades de la Adicción en un esfuerzo por representar a el hecho de que la enfermedad en cuestión es más amplio que los problemas químicos exógenos. A pesar de este progreso avanzado por tales organizaciones y los mejores expertos en el campo, la APA desaprobó explícitamente tanto la palabra «adicción» como la categoría de «adicciones de comportamiento» en el DSM-5. Solo se puede especular sobre si la APA requiere evidencia más estricta para el reconocimiento de trastornos médicos que un grupo de especialidad como el ASAM, o si existe otro estándar, motivo o problema no dicho en juego.
El apoyo adicional a la conclusión de la política social surge cuando se analiza a través de la lente de la decisión de evitar el uso del término «adicción», que ASAM identifica como una enfermedad cerebral. El DSM-5 reconoce el papel de la neurociencia, pero insiste en un sistema de clasificación que identifica los trastornos de conducta, incluidos algunos, pero negando otros. Aunque se propuso su inclusión en el DSM-5, la adicción fue rechazada. Charles O’Brien, (presidente del Grupo de Trabajo de Trastornos por Uso de Sustancias del DSM-5) publicó un artículo que apoya la decisión.
Algunos miembros del grupo de trabajo votaron a favor de un retorno al uso de la palabra «adicción» porque la palabra se ha vuelto tan común en los últimos años y no parece peyorativa para ellos. Los medios de comunicación tienen historias sobre la «adicción al petróleo» y las mujeres visten camisetas adornadas con «adicción al rosa» o a las compras, etc. Por supuesto, las connotaciones de las palabras cambian con el tiempo y la cultura; reconocemos que no hay estudios actuales que puedan ser citados sobre si la elección de las etiquetas podría ser peyorativa. Debido a que algunos científicos siguen oponiéndose al uso de la palabra «adicción», propusimos un compromiso. La etiqueta propuesta en el DSM-V ahora se denomina «trastorno por uso de sustancias» (O’Brien, 2011, pp. 866–867)
En retrospectiva, esta declaración indica que la presión de los pares y del público es la motivación potencial para que el Grupo de Trabajo DSM-5 se aleje de la adicción al término médico. Más allá de la ironía de que esta declaración se publicó en la revista académica llamada Adicción, esta decisión y la declaración son inconsistentes con la afirmación anterior de O’Brien de que el comité de Abuso de Sustancias del DSM-III-R cometió un «grave error» con su decisión de omitir la adicción como una categoría de diagnóstico en el DSM-III-R (O’Brien, Volkow, & Li, 2006). En ese artículo, los autores declararon: “En el caso de los trastornos por uso de sustancias, el mundo médico necesita drásticamente un cambio en el etiquetado. Adicción es una palabra perfectamente aceptable».
Otros ejemplos de la influencia de las políticas sociales en el libro se pueden ver en forma longitudinal, como la adición y posterior eliminación de la homosexualidad como un trastorno mental en la década de 1970, a nuevos escándalos, como el recibo de psiquiatras infantiles Joseph Biederman de $ 1.6 millones de las compañías farmacéuticas. a finales de los 90 / principios de los 2000 para promover el diagnóstico del trastorno bipolar infantil y alentar los medicamentos para su tratamiento. En su libro que documenta el desarrollo del DSM-5, Greenburg (2013) destacó las interacciones cada vez más negativas entre el liderazgo del Grupo de Trabajo del DSM-5 y Robert Spitzer, el presidente del Grupo de Trabajo del DSM-III (a quien se le negó el acceso al comité del DSM-5 actas de reuniones a pesar de sus reclamos públicos de transparencia).
Allen Frances, presidente del Grupo de Trabajo DSM-IV, también ha sido un crítico abierto del DSM-5 (Frances, 2012a, 2012b, 2013), acusando repetidamente a la APA de «inflación diagnóstica», «imperialismo diagnóstico» y reclamos La APA tiene un control monopolístico injusto sobre el desarrollo y la formalización de la nosología de diagnóstico y los conjuntos de criterios (Frances, 2012a). En apoyo de sus declaraciones, Frances ha hecho referencia a las preocupaciones formales expresadas por la Asociación Americana de Consejería, la Sociedad Británica de Psicología (BPS) y las 16 divisiones de la Asociación Americana de Psicología (carta de preocupación unificada). Lamentablemente, ninguna de estas cartas de inquietud formales dio lugar a cambios en el desarrollo o resultado del DSM-5. Frances también hizo referencia a las llamadas internacionales para un boicot al DSM-5 desde Australia, Inglaterra, Francia, Italia y España. Una controversia de particular interés es la renuncia pública de dos miembros del Grupo de Trabajo de Trastornos de Personalidad del DSM-5, quienes expresaron sus razones en un correo electrónico enviado al presidente del Grupo de Trabajo del DSM-IV (Frances, 2012b):
El Grupo de Trabajo sobre Trastornos de la Personalidad … demostró una incapacidad para responder a una retroalimentación constructiva tanto dentro del Grupo de Trabajo como de los muchos expertos en el campo … Al principio del proceso del DSM-5, desarrollamos grandes preocupaciones … No renunciamos antes porque continuamos para atesorar la esperanza de que eventualmente la ciencia y el sentido común prevalecerían … se hizo evidente que no iba a suceder … La propuesta muestra una indiferencia realmente impresionante para la evidencia. Los aspectos importantes de la propuesta carecen de un soporte probatorio razonable de confiabilidad y validez. Esto crea la situación insostenible de que el Grupo de Trabajo avanza en un modelo taxonómico que, según se ha reconocido en un artículo publicado, es inconsistente con la evidencia … Por estas y otras razones, sentimos que el único curso de acción honesto era renunciar al Grupo de Trabajo. (Verheul & Livesley, citado por Frances, 2012b)
Téngase en cuenta que estos autores no se refieren específicamente a la disfunción dentro del Grupo de Trabajo de Trastornos por Uso de Sustancias, sin embargo, sus declaraciones pueden tomarse como indicadores teóricos de la disfunción general y la posible falta de integridad científica en todo el proceso de desarrollo del DSM-5.
Una posible explicación final para los problemas anteriores puede ser tan simple como una mala investigación, lógica y edición. Parte de la lógica de la APA es ilógica. Por ejemplo, declararon que no había suficiente investigación para incluir IA, pero que había suficiente investigación para incluir IGD. Luego citaron principalmente la investigación en IA para apoyar esta posición. Esto es algo así como rechazar A, reclamar A ≠ B donde B es un subconjunto de A y legitimar a B. Igualmente ilógico e inconsistente es el hecho de que APA reconoció y negó a la vez la adicción como un concepto médico, así como «adicciones conductuales” como una categoría válida dentro del espectro de trastornos adictivos, y lo hizo oblicuamente dentro de su capítulo sobre Trastornos relacionados con sustancias y adicciones. Además, incluyen IGD y GD.
Quizás el ejemplo más inesperado sea la declaración hecha por la APA en la sección «Factores de riesgo y pronóstico» del diagnóstico de IGD en el DSM-5 «Genética y fisiológica: los varones adolescentes parecen tener un mayor riesgo de desarrollar un trastorno de los juegos de Internet, y se ha especulado que los antecedentes ambientales y / o genéticos de Asia son otro factor de riesgo, pero esto no está claro”. De alguna manera, ¡la APA logró declarar explícitamente que los asiáticos pueden estar genéticamente predispuestos a desarrollar IGD! De los cientos de artículos revisados por el segundo autor de este artículo, tal afirmación nunca se ha dado a conocer, y mucho menos se ha declarado explícitamente. Quizás la APA pretendía hacer referencia a la cultura asiática como un factor de riesgo ambiental más que como un factor de riesgo genético. Desafortunadamente, el único factor de riesgo ambiental que se menciona es: «La disponibilidad de computadoras con conexión a Internet [sic] permite el acceso a los tipos de juegos con los que se asocia con mayor frecuencia el trastorno de los juegos de Internet» (APA, 2013, p. 797). En conjunto, la APA postuló formalmente que los adolescentes varones asiáticos con computadoras conectadas a Internet son las personas más propensas a desarrollar este trastorno. Esto es una vergüenza para todo el campo de la salud mental, y uno espera una mejor edición de una publicación que cuesta $ 25 millones para producir. Esto en sí mismo sirve como evidencia de facto de una pobre investigación y / o edición.
La APA (2013, p. 783) describió formalmente los beneficios de un diagnóstico que se incluye en la Sección III del DSM en la introducción al capítulo «Condiciones para estudio adicional», en el que indicaron: «Se presentan los conjuntos de criterios propuestos.
para las condiciones en las que se alienta la investigación futura … y están destinadas a proporcionar un lenguaje común para los investigadores y clínicos interesados en estudiar estos trastornos «. Este lenguaje común reconoció a la APA como necesario para la transición de IGD de la Sección III a la Sección II de DSM se necesita desesperadamente para todas las variantes de IA, específicamente para capturar los crecientes problemas relacionados con el uso de Internet para juegos de azar, pornografía, actos sexuales, etc. Los investigadores que investigan la IGD deben seguir el modelo presentado por algunos investigadores asiáticos de reconocer explícitamente la IGD como un subtipo de IA, en lugar de simplemente aceptar el intento de la APA de seleccionar un subtipo del trastorno más grande (Potenza, 2014). Es esencial tener en cuenta la definición de adicción de ASAM que sostiene que la enfermedad de la adicción es primaria y crónica, ya que implica recompensa cerebral, motivación, memoria y circuitos relacionados. Puede manifestarse en términos de búsqueda patológica de recompensa y / o alivio con el uso de sustancias y otros comportamientos. La investigación futura es particularmente necesaria para la API, ya que la lógica dicta que el uso excesivo compulsivo de la pornografía es un ejemplo de adicción al comportamiento. La misma cantidad de investigación que se lleva a cabo en IGD también se requiere en el caso de IPA. Se necesita una evaluación más holística en la investigación y el tratamiento que incluye todos los aspectos de la adicción que son un reflejo de los problemas en la función cerebral, en lugar de centrarse en comportamientos específicos a expensas de excluir los problemas que pueden seguir siendo un problema para los individuos afectados.
Si bien la adicción relacionada con internet se ha reconocido fuera de los Estados Unidos con un lenguaje tan grave como «crisis de salud pública», la APA parece negar abiertamente su existencia, al tiempo que reconoce un solo subtipo. Por ejemplo, el reconocimiento por parte de la APA de que solo los videojuegos en Internet son potencialmente adictivos contribuye al problema que pretenden proporcionar orientación para resolverlos. En otras palabras, sin reconocimiento en el DSM, la evaluación general y el tratamiento de la IA y sus diversos subtipos tendrán un acceso limitado a los fondos necesarios para proporcionar resultados de investigación suficientes para establecer la validez y el tratamiento adecuado de los trastornos adictivos que se incluyen en el marco general. paraguas de la enfermedad de la adicción. Como tal, la APA ha creado actualmente un círculo innecesariamente difícil de ingresar.
Ko et al. (2014) publicaron un estudio en el que validaron la precisión diagnóstica de los criterios del DSM-5 para IGD (aunque sugirieron agregar el deseo como un elemento adicional). En su conclusión, estos autores declararon que no es práctico definir cada actividad adictiva (pornografía, redes sociales, etc.) en Internet como un trastorno distinto, en oposición a los subtipos de un trastorno más grande. Desafortunadamente, esto es exactamente lo que la APA ha propuesto:
El uso excesivo de Internet que no implique jugar juegos en línea (por ejemplo, el uso excesivo de redes sociales, como Facebook; ver pornografía en línea) no se considera análogo al trastorno de los juegos de Internet, y la investigación futura sobre otros usos excesivos de Internet debe seguir pautas similares a las sugeridas aquí.
ASAM declaró claramente que todos los aspectos de la adicción se refieren a problemas comunes en los circuitos cerebrales, no a las diferencias en la (s) sustancia (s) o contenido o comportamiento (s) (ASAM, 2011). Por lo tanto, con base en la opinión de expertos y los hallazgos revisados en Love et al. (2015), es ilógico que la APA desapruebe explícitamente algunos comportamientos patológicos de Internet mientras permite otros. Esta decisión y declaración no es lógica ni coherente con la evidencia científica existente y emergente. Según esta lógica, la visualización excesiva de la propiedad intelectual y los juegos en Internet son sustancialmente diferentes, a pesar de la superposición sustancial en la activación del sistema de recompensa del cerebro, y a pesar del potencial para la exhibición de comportamientos psicosociales similares y consecuencias psicosociales. Esto es, inconsistente desde el punto de vista biológico y de comportamiento” (Hilton, 2013).
La mala interpretación de la neurociencia de la adicción se puede ver en la sección de Características de diagnóstico del DSM-5 para la IGD, en la que se hace referencia a los aspectos de grupo y equipo como características clave del trastorno. Según esta lógica, el abuso de sustancias en un bar o en una fiesta puede constituir abuso de sustancias, pero el abuso de sustancias cuando está solo no lo hace. Para hacer una analogía relacionada con Internet, esta lógica dicta que alguien que juega a World of Warcraft en exceso es adicto, pero que alguien que juega a Candy Crush en exceso no lo es. La desestimación de la ciencia establecida por parte de la APA a favor de las opiniones es lo que parece haber llevado al NIMH a dejar de basar la investigación en las categorías de DSM y, en cambio, a sustituir sus propias normas de investigación más científicas (Insel et al., 2013).
Instamos a las comunidades de investigación y tratamiento a ser más rigurosas y consistentes para que las poblaciones afectadas por la adicción reciban evaluaciones mejores y más holísticas.
eso guiaría un mejor tratamiento y seguimiento en el contexto de la adicción como una enfermedad crónica en lugar del enfoque actual en uno o más trastornos de la conducta que pueden o no ser controlados, mientras que otros aspectos de la adicción permanecen sin abordar.
Referencias de los autores: Raju Hajelaa & Todd Loveb, 2017 a American Society of Addiction Medicine, Diagnostic and Descriptive Terminology Action Group, Chevy Chase, Maryland; b Society for the Advancement of Sexual Health, Ardmore, Pennsylvania
Referencias
American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders, fifth edition (DSM-V). Arlington, VA: Author.
Frances, A. (2013). Saving normal: An insider’s revolt against out-of-control psychiatric diagnosis, DSM-5, big pharma and the medicalization of ordinary life. New York, NY: HarperCollins.
Fu, K. W., Chan, W. S., Wong, P. W., & Yip, P. S. (2010). Internet addiction: Prevalence, discriminant validity and correlates among adolescents in Hong Kong. British Journal of Psychiatry, 196, 6.
Han, D. H., Hwang, J. W., & Renshaw, P. F . (2010). Bupropion sustained release treatmentdecreases craving for video games and cue-induced brain activity in patients with Internet video game addiction. Experimental and Clinical Psychopharmacology, 18, 297–304. doi:10.1037/a0020023
Karaiskos, D., Tzavellas, E., Balta, G., & Paparrigopoulos, T. (2010). P02–232-Social network addiction: A new clinical disorder? European Psychiatry, 25, 855.
Kim, M. G., & Kim, J. (2010). Cross-validation of reliability, convergent and discriminant validity for the problematic online game use scale. Computers in Human Behavior, 26, 389–398.
Kim, S. H., Baik, S. H., Park, C. S., Kim, S. J., Choi, S. W., & Kim, S. E. (2011). Reduced striatal dopamine D2 receptors in people with Internet addiction. Neuroreport, 22, 407–411. doi:10.1097/WNR.0b013e328346e16e
King, D. L., & Delfabbro, P. H. (2013). Issues for DSM-5: Video-gaming disorder? Australian and New Zealand Journal of Psychiatry, 47, 20–22.
Kittinger, R., Correia, C. J., & Irons, J. G. (2012). Relationship between Facebook use and problematic Internet use among college students. Cyberpsychology Behavior Social Network, 15, 324–327. doi:10.1089/cyber.2010.0410
Ko, C. H., Yen, J. Y., Chen, C. C., Chen, S. H., & Yen, C. F. (2005). Proposed diagnostic criterio of Internet addiction for adolescents. Journal of Nervous and Mental Disorders, 193, 728–733. doi:10.1097/01.nmd.0000185891.13719.54
Ko, C.-H., Yen, J.-Y., Chen, S.-H., Wang, P.-W ., Chen, C.-S., & Yen, C.-F. (2014). Evaluation of the diagnostic criteria of Internet gaming disorder in the DSM-5 among young adults in Taiwan. Journal of Psychiatric Research, 53, 103–110.
Koc, M., & Gulyagci, S. (2013). Facebook addiction among Turkish college students: The role of psychological health, demographic, and usage characteristics. Cyberpsychology, Behavior, and Social Networking, 16, 279–284. doi:10.1089/cyber.2012.0249
Meerkerk, G.-J., Eijnden, R. J. V. D., & Garretsen, H. F. (2006). Predicting compulsive Internet use: It’s all about sex! CyberPsychology & Behavior, 9, 95–103. doi:10.1089/cpb.2006.9.95
Milosevic-Đorđevic, J. S., & Zezelj, I. L. (2014). Psychological predictors of addictive social networking sites use: The case of Serbia. Computers in Human Behavior, 32, 229–234.
O’Brien, C. P., Volkow, N., & Li, T. K. (2006). What’s in a word? Addiction versus dependence in DSM-V. American Journal of Psychiatry, 163, 764–765. doi:10.1176/appi.ajp.163.5.764
Potenza, M. N. (2006). Should addictive disorders include nonsubstancerelated conditions? Addiction, 101, 142–151. doi:10.1111/j.1360-0443.2006.01591.x
Rosen, L. D., Whaling, K., Rab, S., Carrier, L. M., & Cheever, N. A. (2013). Is Facebook creating “iDisorders”? The link between clinical symptoms of psychiatric disorders and technology use, attitudes and anxiety. Computers in Human Behavior, 29, 1243–1254. doi:10.1016/j. chb.2012.11.012
Salehan, M., & Negahban, A. (2013). Social networking on smartphones: When mobile phones become addictive. Computers in Human Behavior, 29, 2632–2639.
Shek, D. T., Tang, V. M., & Lo, C. Y. (2009). Evaluation of an Internet addiction treatment program for Chinese adolescents in Hong Kong. Adolescence, 44, 359–373.
Starcevic, V. (2013). Is Internet addiction a useful concept? Australian and New Zealand Journal of Psychiatry, 47, 16–19.
Stewart, C. S. (2010). Obsessed with the Internet: A tale from China. Wired, 01/13/2010.
Tsitsika, A. E. C. A. L., Janikian, M., Freskou, A., Marangou, E., … Kafetzis, D. (2011). Determinants of Internet addiction among adolescents: A case-control study. Scientific World Journal, 11, 8.
Turel, O., He, Q., Xue, G., Xiao, L., & Bechara, A. (2014). Examination of neural systems subserving Facebook “addiction.” Psychological Report, 115, 675–695. doi:10.2466/18. PR0.115c31z8
Van Rooij, A. J., Schoenmakers, T. M., Vermulst, A. A., Van den Eijnden, R. J., & Van de Mheen, D. (2011). Online video game addiction: Identification of addicted adolescent gamers. Addiction, 106, 205–212. doi:10.1111/j.1360-0443.2010.03104.x
Weinstein, A., & Lejoyeux, M. (2010). Internet addiction or excessive Internet use. American Journal of Drug and Alcohol Abuse, 36, 6.
Weiss, R., & Samenow, C. P. (2010). Smart phones, social networking, sexting and problematic sexual behaviors—a call for research. Sexual Addiction & Compulsivity, 17, 241–246.
Widyanto, L., Griffiths, M. D., & Brunsden, V. (2011). A psychometric comparison of the Internet Addiction Test, the Internet-Related Problem Scale, and self-diagnosis. Cyberpsychology, Behavior, and Social Networking, 14, 141–149. doi:10.1089/cyber.2010.0151
Young, K. (1998). Internet addiction: The emergence of a new clinical disorder. CyberPsychology & Behavior, 1, 237–244.
Zhou, Y., Lin, F. C., Du, Y. S., Qin, L. D., Zhao, Z. M., Xu, J. R., & Lei, H. (2011). Gray matter abnormalities in Internet addiction: A voxel-based morphometry study. European Journal of Radiology, 79, 92–95. doi:10.1016/j.ejrad.2009.10.025